Arq. Bras. Oftalmol. 2010; 73 (3): 10.1590/S0004-27492010000300015
Total: 3526
Luiza Caye1; Karin Scheid2; Melissa Manfroi Dal Pizzol3; Roberto Freda4
DOI: 10.1590/S0004-27492010000300015
RESUMO
A síndrome KID é uma displasia ectodérmica congênita rara, caracterizada pela associação de lesões hiperceratóticas de pele, surdez neurossensorial e ceratite. O comprometimento ocular ocorre em 95% dos portadores da síndrome. Embora as manifestações dermatológicas tenham sido bastante estudadas, sabe-se pouco sobre o manejo e o prognóstico do acometimento oftalmológico. Até o momento, o tratamento ocular da síndrome KID inclui tratamento sintomático, não havendo na literatura drogas ou tratamento cirúrgico que possam modificar o curso e prognóstico da doença. Neste relato de caso, descrevemos os achados oftalmológicos de um portador de síndrome KID e o uso de bevacizumab (Avastin®) subconjuntival como sugestão para o tratamento da neovascularização corneana. Apesar da ausência de melhora e da raridade da doença, os autores acreditam que a utilização do bevacizumab subconjuntival possa ser útil para o tratamento da neovascularização corneana que se estabelece na síndrome KID e incentivam os pesquisadores a se aprofundar neste tema.
Descritores: Síndrome; Surdez; Displasia ectodérmica; Ceratite; Neovascularização corneana; Bevacizumab; Relatos de casos
ABSTRACT
KID syndrome is a congenital ectodermal dysplasia characterized by the association of keratitis, hyperkeratotic skin lesions and neurosensorial hearing loss. Ocular involvement occurs in 95% of patients. Although KID syndrome cutaneous manifestations have been studied in-depth, the treatment and prognosis of ophthalmic impairment have not been described in detail. At present, the treatment of the ocular damage caused by the syndrome is symptomatic and there are no studies defining a treatment that could change the disease course. In this case, ophthalmologic findings of a patient with KID syndrome and the use of subconjunctival bevacizumab to treat corneal neovascularization are described. In spite of the absence of improvement in this patient and the few reports of this disease, aditional studies with bevacizumab to treat corneal deep neovascularization are suggested.
Keywords: Syndrome; Deafness; Ectodermal dysplasia; Keratitis; Corneal neovascularization; Bevacizumab; Case reports
INTRODUÇÃO
A primeira descrição de doença inflamatória corneana progressiva, eritroderma difuso hiperceratótico e surdez neurossensorial foi feita por Burns, em 1915. Posteriormente, em 1981, estas alterações foram reconhecidas como uma displasia ectodérmica congênita rara, a síndrome KID - ceratite (K), ictiose (I) e surdez neurossensorial (D)(1). Outros achados incluem alopécia, distrofia de unhas, anormalidades dentárias, maior risco para desenvolvimento de carcinoma de células escamosas e maior suscetibilidade para infecções bacterianas e fúngicas(2-4).
A maioria dos casos de síndrome KID é esporádica. A apresentação familiar, entretanto, também é descrita, sugerindo que pode haver herança genética ainda não esclarecida(5). As manifestações dermatológicas da síndrome têm sido bastante estudadas. Por outro lado, sabe-se pouco sobre o manejo e o prognóstico do acometimento oftalmológico. Neste relato de caso, são descritos os achados oftalmológicos de portador de síndrome KID, no qual foi usado bevacizumab (Avastin®) subconjuntival para o tratamento da neovascularização corneana.
RELATO DE CASO
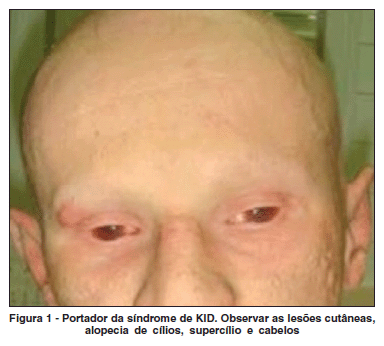
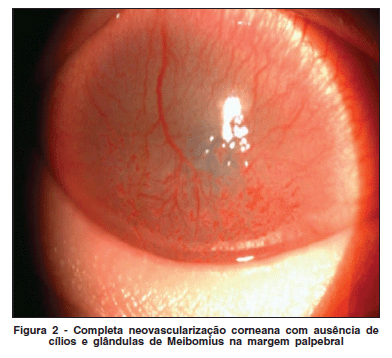
Paciente masculino, 21 anos, com diagnóstico de síndrome KID, foi encaminhado ao serviço de Oftalmologia do Hospital Banco de Olhos de Porto Alegre. Relatava opacidade corneana bilateral, iniciada alguns meses após o nascimento. Ao exame, apresentava lesões hiperceratóticas difusas na pele, anidrose, alopécia de cabelos, sobrancelhas e cílios (Figura 1), surdez neurossensorial e dificuldade na fala. A acuidade visual era de conta dedos a 1,5 metros em ambos os olhos. A biomicroscopia revelava hiperemia conjuntival média, neovascularização da superfície corneana (superficial e profunda) em 360º (Figura 2) e redução marcante do menisco lacrimal em ambos os olhos. Os valores do teste de Schirmer foram de 5 mm no olho direito e 6, no esquerdo. A fundoscopia não foi possível, devido à neovascularização corneana. A ecografia ocular foi normal. Foi iniciado tratamento sintomático, com lubrificação intensiva usando colírio de carboximetilcelulose sódica 1% de 1/1 h e gel de ácido poliacrílico 0,3% à noite. Na tentativa de reduzir a neovascularização corneana, foi realizada uma injeção subconjuntival a 1 mm do limbo de 0,1 ml de bevacizumab 2,5 mg/ml nos quatro quadrantes de ambos os olhos. Paciente apresentou melhora parcial dos sintomas de olho seco, sendo que não foi observada melhora dos neovasos após aplicação da medicação.
DISCUSSÃO
A síndrome KID é uma anomalia congênita rara de tecidos ectodérmicos, de incidência desconhecida, havendo cerca de 100 casos publicados no mundo. Usualmente, manifesta-se como casos isolados, acometendo igualmente ambos os sexos(6). Caracteriza-se pela associação de lesões hiperceratóticas de pele, surdez neurossensorial profunda e ceratite(6). Estudos descrevem que 95% dos portadores da síndrome possuem comprometimento ocular. Fotofobia, irritação ocular, lacrimejamento e baixa acuidade visual decorrente da neovascularização corneana são os principais sintomas associados(6). As manifestações oculares da síndrome KID ocorrem geralmente na infância, após o surgimento das alterações cutâneas e auditivas. Portanto, o acrônimo KID pode não estar completo nas fases precoces da doença(7).
O mecanismo da doença corneana não parece ser inflamatório, como implica o termo "ceratite", mas resultante de um distúrbio ectodérmico generalizado, com atrofia ou ausência das glândulas de Meibomius, queratinização dos ductos das glândulas lacrimais e deficiência de células basais limbares. Consequentemente, há desenvolvimento de olho seco grave e formação de pannus corneano(6,8-9).
Mutações no gene GJB2 que codifica a conexina 26 - componente das "gap junctions" das células epiteliais - recentemente têm sido descritas em pacientes com síndrome KID. Embora a distribuição da conexina 26 na córnea ainda não esteja bem definida, disfunções nas "gap junctions" do epitélio corneano podem ser responsáveis por alterações da superfície ocular(10-12).
O tratamento ocular na síndrome KID é sintomático, não havendo ainda descrição de drogas ou tratamento cirúrgico que possam modificar o curso e prognóstico dessa doença. Lágrimas artificiais e agentes anti-inflamatórios como corticóides tópicos e ciclosporina A têm sido utilizados, com respostas variáveis(6). Procedimentos cirúrgicos incluindo ceratectomia superficial, transplante alógeno de limbo, uso de membrana amniótica e ceratoplastia lamelar e penetrante têm sido realizados sem sucesso(8). Recentemente, tem-se comprovado aumento da produção do fator de crescimento endotelial vascular (VEGF) em doenças inflamatórias corneanas e da superfície ocular, tanto em modelo humano, quanto em animais, sendo esse fator diretamente envolvido na gênese da neovascularização corneana. O bevacizumab é um anticorpo monoclonal humano que age contra o VEGF solúvel, inibindo sua ligação ao receptor, impedindo a proliferação de células endoteliais e formação vascular. O uso de injeção subconjuntival de bevacizumab no tratamento da neovascularização corneana em pacientes com síndrome KID ainda não havia sido sugerido na literatura. A falta de regressão da neovascularização no caso descrito pode ser atribuída à cronicidade do caso e ao fato de que o insulto inflamatório decorrente do olho seco grave permaneça presente, apesar do tratamento instituído.
A neovascularização corneana com consequente perda da transparência é a complicação ocular de pior prognóstico visual da síndrome de KID. A utilização do bevacizumab subconjuntival em fases precoces da doença poderia impedir a neovascularização corneana.
Embora o prognóstico geral da síndrome KID pareça ser bom, é necessário acompanhamento multidisciplinar precoce e a longo prazo dos indivíduos acometidos. Deve-se atentar para a maior suscetibilidade a infecções e risco aumentado de desenvolvimento de tumores malignos, particularmente carcinoma de células escamosas(2-4,8). A lubrificação intensa da superfície ocular vai reduzir as complicações.
REFERÊNCIAS
1. Skinner BA, Greist MC, Norins AL. The keratitis, ichthyosis, and deafness (KID) syndrome. Arch Dermatol. 1981;117(5):285-9.
2. Langer K, Konrad K, Wolff K. Keratitis, ichthyosis and deafness (KID)-syndrome: report of three cases and a review of the literature. Br J Dermatol. 1990;122(5):689-97.
3. Gilliam A, Williams ML. Fatal septicemia in an infant with keratitis, ichthyosis and deafness (KID) syndrome. Pediatr Dermatol. 2002;19(3):232-6.
4. Wilson GN, Squires RH Jr, Weinberg AG. Keratitis, hepatitis, ichthyosis and deafness: report and review of KID syndrome. Am J Med Genet. 1991;40(3):225-9.
5. Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, Man SY, Bodemer C, Prins C, et al. Keratitis-ichthyosis-deafness syndrome: disease expression and espectrum of connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol. 2007; 156(5):1015-9.
6. Gómez-Faíña P, Ruiz-Viñals AT, Buil-Calvo JA, España-Albelda A, Pazos-López M, Castilla-Céspedes M. [Patient with severe corneal disease in KID syndrome]. Arch Soc Esp Oftalmol. 2006;81(4):225-7. Spanish.
7. Caceres-Rios H, Tamayo-Sanchez L, Duran-Mckinster C, de la Luz Orozco M, Ruiz-Maldonado R. Keratitis, ichthyosis, and deafness (KID syndrome): review of the literature and proposal of a new terminology. Pediatr Dermatol. 1996; 13(2):105-13. Comment in: Pediatr Dermatol. 1996;13(2):154-7. Pediatr Dermatol. 2000;17(3):244-5.
8. Messmer EM, Kenyon KR, Rittinger O, Janecke AR, Kampik A. Ocular manifestations of keratitis-ichthyosis-deafness (KID) syndrome. Ophthalmology. 2005;112(2):e1-6.
9. Sonoda S, Uchino E, Sonoda KH, Yotsumoto S, Uchio E, Isashiki Y, Sakamoto T. Two patients with severe corneal disease in KID syndrome. Am J Ophthalmol. 2004;137(1):181-3.
10. Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynänen M, et al. Missence mutatios in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70(5):1341-8.
11. Alvarez A, del Castilho I, Pera A, Villamar M, Moreno-Pelayo MA, Moreno F, et al. De novo mutation in the gene encoding connexin-26 (GJB2) in a sporadic case of keratitis-ichthyosis-deafness (KID) syndrome. Am J Med Genet. 2003; 117A(1):89-91.
12. Yotsumoto S, Hashiguchi T, Chen X, Ohtake N, Tomitaka A, Akamatsu H, et al. Novel mutations in GJB2 encoding connexin-26 in Japanese patients with keratitis-ichthyosis-deafness syndrome. Br J Dermatol. 2003;148(4):649-53.
Endereço para correspondência:
Melissa Manfroi Dal Pizzol
Rua Casemiro de Abreu, 908/201 - Porto Alegre (RS)
CEP 90420-000
E-mail: [email protected]
Recebido para publicação em 20.07.2009
Última versão recebida em 23.09.2009
Aprovação em 25.09.2009
Trabalho realizado no Hospital Banco de Olhos de Porto Alegre - Porto Alegre (RS) - Brasil.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.