Arq. Bras. Oftalmol. 2009; 72 (4): 10.1590/S0004-27492009000400026
Total: 2723
Monique Côco1; Sang Won Han3; Juliana Maria Ferraz Sallum
DOI: 10.1590/S0004-27492009000400026
RESUMO
As distrofias hereditárias de retina abrangem um amplo número de doenças caracterizadas por lenta e progressiva degeneração da retina. São o resultado de mutações em genes expressos em fotorreceptores e no epitélio pigmentado da retina. A herança pode ser autossômica dominante, autossômica recessiva, ligada ao X recessiva, digênica ou herança mitocondrial. Atualmente não há tratamento para essas doenças e os pacientes convivem com a perda progressiva da visão. O aconselhamento genético e o suporte para reabilitação têm indicação nestes casos. Pesquisas envolvendo a base molecular e genética dessas doenças está continuamente em expansão e ampliam as perspectivas para novas formas de tratamento. Dessa forma, a terapia gênica, que consiste na inserção de material genético exógeno em células de um indivíduo com finalidade terapêutica, tem sido a principal forma de tratamento para as distrofias hereditárias de retina. O olho é um órgão peculiar para a terapia gênica, pois é anatomicamente dividido em compartimentos, imunologicamente privilegiado e com meios transparentes. A maioria das doenças oculares tem defeitos em genes conhecidos. Além disso, há modelo animal bem caracterizado para algumas condições. Propostas para pesquisa clínica em terapia gênica nas degenerações retinianas hereditárias com defeito no gene RPE65, recentemente tiveram aprovação ética e os resultados preliminares obtidos trouxeram grandes expectativas na melhora da qualidade de vida dos pacientes.
Descritores: Retina; Oftalmopatias hereditárias; Terapia de genes; Vetores genéticos; Técnicas de transferência de genes
ABSTRACT
The inherited retinal dystrophies comprise a large number of disorders characterized by a slow and progressive retinal degeneration. They are the result of mutations in genes that express in either the photoreceptor cells or the retinal pigment epithelium. The mode of inheritance can be autosomal dominant, autosomal recessive, X linked recessive, digenic or mitochondrial DNA inherited. At the moment, there is no treatment for these conditions and the patients can expect a progressive loss of vision. Accurate genetic counseling and support for rehabilitation are indicated. Research into the molecular and genetic basis of disease is continually expanding and improving the prospects for rational treatments. In this way, gene therapy, defined as the introduction of exogenous genetic material into human cells for therapeutic purposes, may ultimately offer the greatest treatment for the inherited retinal dystrophies. The eye is an attractive target for gene therapy because of its accessibility, immune privilege and translucent media. A number of retinal diseases affecting the eye have known gene defects. Besides, there is a well characterized animal model for many of these conditions. Proposals for clinical trials of gene therapy for inherited retinal degenerations owing to defects in the gene RPE65, have recently received ethical approval and the obtained preliminary results brought large prospects in the improvement on patient's quality of life.
Keywords: Retina; Eye diseases, hereditary; Gene therapy; Genetic vectors; Gene transfer techniques
INTRODUÇÃO
Terapia gênica consiste na inserção de material genético exógeno em células de um indivíduo com finalidade terapêutica.
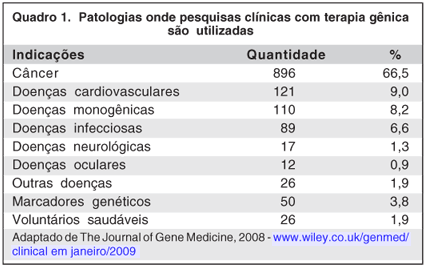
A transferência tem como objetivo recuperar a função de um gene, atribuir uma nova atividade gênica ou potencializar a atividade dos genes ativos. Com estes objetivos, esta terapia pode ser aplicada tanto para doenças genéticas quanto para adquiridas(1-2) (Quadro 1).
O olho é um órgão peculiar para a terapia gênica, pois é anatomicamente dividido em compartimentos (situação ideal para inserção precisa do vetor no tecido de interesse), imunologicamente privilegiado e com meios transparentes. A maioria das doenças oculares tem defeitos em genes conhecidos e tem progressão lenta permitindo maior tempo para tratamento. O RetNet, Retinal Information Network (disponível em http://www.sph. uth.tmc.edu/retnet/), é um banco de dados online sobre os genes da retina. Além disso, há um modelo animal bem caracterizado para algumas condições, facilitando a pesquisa pré-clínica.
Para a terapia gênica em degenerações retinianas é desejável que o gene inserido se expresse por um longo tempo e tenha regulações apropriadas. Existem muitas degenerações retinianas monogênicas e algumas dessas doenças são candidatas à terapia gênica, pois, até o momento, ainda não há outro tratamento.
Há duas principais estratégias de tratamento para degenerações retinianas em fase de experimentação: injeção intraocular de fatores de crescimento e restauração da função por transferência de genes à células(3).
O gene terapêutico pode ser introduzido num indivíduo de duas formas: in vivo ou ex vivo. Na técnica in vivo a introdução do gene ocorre diretamente no organismo, enquanto que na técnica ex vivo, as células são retiradas do indivíduo, modificadas geneticamente e depois reintroduzidas.
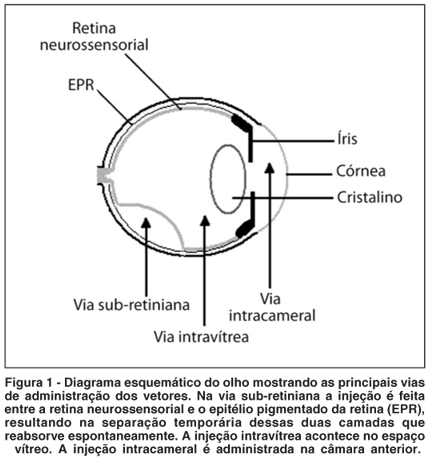
A introdução de genes na retina pode ser realizada por várias vias, tais como: injeção intravítrea, sub-retiniana e intracameral (Figura 1). Injeção intravítrea é um método fácil e relativamente seguro, mas há complicações como hemorragia vítrea, descolamento de retina e endoftalmites. Têm sido mais utilizada para transfectar células ganglionares. Injeção sub-retiniana, com ou sem vitrectomia, que produz um desprendimento local e transitório da retina tem sido o método de escolha para acessar a camada de fotorreceptores e epitélio pigmentado da retina (EPR). Injeção intracameral é mais utilizada para células do segmento anterior do olho (endotélio corneano, epitélio pigmentado da íris, corpo ciliar)(4).
Inicialmente, deve-se definir o cassete de expressão gênica, que é uma sequência de DNA contendo as informações do gene de interesse e dos elementos regulatórios que vão controlar sua expressão(1-2).
Um segundo ponto relevante consiste em se determinar o veículo (ou vetor) que carregará o cassete de expressão até a célula-alvo(5).
O vetor ideal possui algumas características importantes, como: baixa imunogenicidade e citotoxicidade, capacidade de expressar de forma estável e duradoura o transgene, direcionamento para tipos específicos de células ou tecidos, baixo custo, fácil manipulação e produção em larga escala e a possibilidade de regular o tempo de expressão do gene exógeno.
Até o momento não foi possível produzir este vetor ideal, mas, levando em consideração o tecido-alvo, doença, duração do tratamento, aplicação in vivo ou ex vivo, tratamento localizado ou sistêmico, resposta imunológica, tamanho do transgene e interferência de elementos regulatórios, é possível determinar o melhor vetor que carregará o material genético e o introduzirá na célula-alvo, que passará a expressá-lo(4-5).
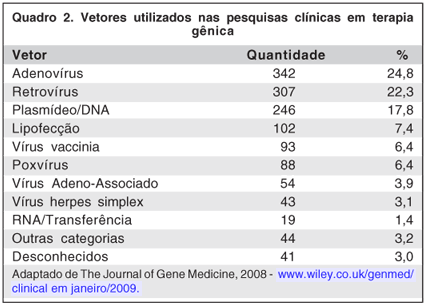
A utilização de vetores virais para a transferência gênica é o método mais comum. Outras formas incluem princípios físicos ou químicos (Quadro 2).
Um método físico bastante conhecido é a eletroporação. Neste sistema, pulsos elétricos alternados são aplicados a células que estão em contato com uma solução de DNA plasmidial. A corrente gerada é capaz de formar poros na superfície celular, facilitando a entrada do material genético nas células(6).
A biobalística ou "gene gun" é outro método físico importante, onde microesferas de ouro ou tungstênio cobertas com DNA são aceleradas por um gás carreador que projeta estas esferas contra células, promovendo a entrada deste DNA no núcleo das células bombardeadas(6).
Os métodos químicos consistem na interação entre um lipídeo catiônico (lipoplexo) ou polímero catiônico (poliplexo) e o DNA, onde a carga positiva do complexo interage com o grupamento fosfato do DNA. A internalização celular ocorre por endocitose e o vetor é degradado no citoplasma deixando a molécula de DNA livre para entrar no núcleo(5-6).
VETORES VIRAIS
Atualmente é o método mais utilizado para terapia gênica devido à alta eficiência de transfecção.
Os vírus são modificados para que fiquem deficientes em replicação e capazes de transferir seu material genético para células-alvo. Isso acontece através da deleção de genes virais envolvidos na proliferação viral e substituição destes pelo gene de interesse. Genes importantes no desenvolvimento da doença também são inativados.
As famílias de vírus mais conhecidas e utilizadas em protocolos de transferência gênica são: os retrovírus, adenovírus, vírus adeno-associado e herpes simples vírus.
Retrovírus
Lentivírus e oncorretrovírus fazem parte da família dos retrovírus. Retrovírus são vírus de RNA fita simples, transcrito para DNA pela transcriptase reversa que o vírus carrega e integrado no genoma da célula infectada. Os lentivírus têm a capacidade de transfectar células quiescentes. Já os oncorretrovírus têm preferência por células em divisão, como tumores oculares. Sua propriedade de integração ao genoma hospedeiro acentua a possibilidade de garantir a expressão estável do transgene.
No entanto, existem problemas como a integração aleatória dos retrovírus no genoma hospedeiro podendo inserir-se no meio de outro gene modificando sua expressão naquela célula. Outro problema de grande importância é a questão de segurança na manipulação de vetores virais baseados em vírus como o HIV(7-8).
Takahashi et al., constataram que o uso do HIV como vetor é útil na transfecção das células fotorreceptoras apenas em degenerações retinianas recessivas. O vetor não produziu efeitos adversos, mostrando ser seguro(7).
Bainbridge et al., descreveram a consequência do uso de lentivírus HIV na transfecção celular. De acordo com esses autores, a injeção sub-retiniana promoveu transfecção eficiente e estável do EPR e baixa transfecção das células fotorreceptoras. Porém, a injeção intravítrea foi ineficiente(8).
Adenovírus
Para transfecção eficiente in vivo foram elaborados vetores adenovirais. Adenovírus são pequenos vírus de DNA dupla fita. Esses vetores conseguem infectar a maioria dos tipos celulares de mamíferos, além de ser possível purificá-los em larga escala. Entretanto, o efeito citotóxico nas células infectadas causa diminuição da expressão do transgene.
Li et al., observaram que após injeção sub-retiniana, houve alta eficiência de transfecção do EPR, mas pouca transfecção das células fotorreceptoras. Observou também, que essas células podem ser melhor transfectadas se estiverem em processo de desenvolvimento ou degeneração. Em ambos os casos, os fotorreceptores são mais acessíveis ao vírus(9).
O problema está no fato de algumas das degenerações retinianas terem início tardio ou o diagnóstico ser confirmado muito tempo após o nascimento.
Bennett et al., demonstraram que os vetores adenovirais podem levar o gene exógeno até as células do espaço sub-retiniano - células fotorreceptoras, células de Muller e células do EPR. A transferência gênica para essas células ocorreu apenas quando o adenovírus foi injetado no espaço sub-retiniano. Observaram também regeneração e reorganização dos segmentos externos dos fotorreceptores no período de duas semanas(10).
Nesse mesmo estudo, Bennett et al., reportaram que a tranfecção da retina neurossensorial em camundongos adultos requer a injeção de altos títulos de adenovírus. Entretanto, há a possibilidade de toxicidade direta das células, especialmente as células adjacentes ao EPR(10).
Vírus Adeno-Associado (AAV)
O genoma do AAV contém uma fita simples de DNA. Nas células humanas o vírus fica latente, sua ativação ocorre na presença de um vírus auxiliador, como o adenovírus ou herpes simples vírus.
O AAV é uma atraente alternativa, pois não está associado a nenhuma doença em humanos. Pode infectar grande variedade de células, é menos imunogênico e transfecta fotorreceptores com mais eficiência que os adenovírus. Por ter tamanho reduzido, o vírus é capaz de penetrar nas camadas da retina mais facilmente.
Os vetores AAV têm como limitações sua difícil preparação em altos títulos, tamanho reduzido do inserto que o vetor consegue acomodar e necessidade de co-infecção das células com o vírus auxiliador para facilitar a conversão da simples fita de DNA em DNA dupla fita.
O primeiro caso mostrando a capacidade dos AAV em transfectar células fotorreceptores foi reportado por Ali et al. Neste estudo, após injeção sub-retiniana de partículas de AAV codificando o gene repórter LacZ em olhos de camundongos, foi demonstrado transfecção de todas as camadas da retina neurossensorial com sucesso. Sendo que a eficiência de transfecção das células fotorreceptoras com AAV foi significativamente maior que a obtida com vetores adenovirais(11).
Jomary et al., demostraram que além da capacidade do AAV em transfectar todas as camadas da retina neurossensorial, também era útil no tratamento das degenerações retinianas, pois, os olhos dos camundongos tratados mostraram aumento da sobrevivência de fotorreceptores e aumento da sensibilidade à luz, medida pela eletrorretinografia(12).
VETORES NÃO VIRAIS
Vetores não virais de transferência gênica são mais fáceis de manipulação, produção e purificação em larga escala do que os vetores virais.
Embora estudos demonstrem que vetores não virais sejam menos eficientes, possuem a vantagem de baixa toxicidade e imunogenicidade. Também podem ser usados para aumentar a eficiência da transfecção por vetores virais como descrito por Hodgson, Solaiman, que obtiveram maior eficiência de transfecção retroviral utilizando um lipossomo catiônico(13).
Recentemente, estudos utilizando nanopartículas de DNA mostraram ser uma boa alternativa para o uso de lipossomos, já que são mais eficientes na transfecção, causam menos toxicidade, provando ser um útil veículo para DNA. A nanopartícula contém um segmento de DNA ou RNA empacotado em um polímero catiônico. Essas partículas penetram pela superfície celular e atingem o núcleo em curto período de tempo(14).
Farjo et al., observaram que dois dias após injeção intravítrea ou sub-retiniana de nanopartículas contendo o gene repórter proteína verde fluorescente (GFP- Green Fluorescent Protein) em olhos de camundongos adultos houve alta taxa de expressão do gene em vários tecidos oculares. Injeção intravítrea levou a alta expressão de GFP na retina (células ganglionares e camada plexiforme interna) e segmento anterior (córnea, trabeculado e cristalino). Com injeção sub-retiniana houve alta taxa de GFP expresso na camada nuclear externa, EPR e nervo óptico. A nanopartícula não se limitou à área de administração e não houve evidência de inflamação. Dessa forma, as nanopartículas provaram ser úteis para múltiplos tipos de doenças oculares(15).
TERAPIA GÊNICA VISANDO NEUROPROTEÇÃO
A observação que a apoptose possa ser manipulada por fatores de crescimento sugeriu uma alternativa terapêutica para muitas doenças degenerativas, dentre elas as doenças degenerativas do olho(16).
O estudo de Fakrorovich et al., foi a primeira indicação de viabilidade desta técnica. Injeções de bFGF (Fibroblast Growth Factor) no olho de ratos, resultaram em longo tempo de sobrevivência dos fotorreceptores. A desvantagem do bFGF é sua grande especificidade, sendo influenciado por diferentes tipos celulares(17). Borhani et al., estudaram a quantidade do fator bFGF por injeção intravítrea e seus efeitos. Constataram, por exemplo, que em coelhos, altas doses desse fator poderiam causar catarata, tração vítrea e vitreorretinopatia proliferativa(18).
Estudo realizado por Cayouette et al., demonstrou que injeção intravítrea de CNTF (Ciliary Neurotrophic Factor) aumenta a sobrevivência dos fotorreceptores e a amplitude da resposta de bastonetes à luz na eletrorretinografia. Esses efeitos foram acompanhados por significantes mudanças na morfologia dos segmentos dos fotorreceptores e elevação do conteúdo total de rodopsina na retina(19).
Essa forma de tratamento tem a vantagem de ser efetiva em doenças com mutações genéticas desconhecidas. Possui a desvantagem do baixo tempo de efeito do fator neurotrófico, durando até um mês em animais.
Repetidas injeções intravítreas da proteína apresentam dificuldades e riscos como descolamento de retina ou endoftalmites.
Para evitar esse problema, a Neurotech Pharmaceuticals desenvolveu uma técnica onde células do EPR são encapsuladas em um pequeno dispositivo que lentamente libera CNTF intraocular. Esse processo foi usado pelo grupo de Aguirre em cães com retinose pigmentar por mutação no gene PDE6B e provou ser eficiente, prevenindo a grave perda de fotorreceptores(20).
Atualmente pesquisas clínicas utilizando o produto Neurotech estão em fase II/III para o tratamento de doenças relacionadas à retinose pigmentar.
RETINOSE PIGMENTAR
O termo retinose pigmentar (RP) é usado para um grupo de distrofias retinianas progressivas e hereditárias. É caracterizada por dificuldade ou ausência de adaptação ao escuro, perda de campo visual periférico, perda da acuidade visual até cegueira total, visão de cores anormal e fotofobia(21). O padrão de herança pode ser autossômico dominante (AD), autossômico recessivo (AR), ligado ao X recessivo ou isolado(22).
Há mais de 40 genes diferentes relacionados com a RP. No gene da rodopsina (RHO, 3q21-q25), que codifica o pigmento fotossensível nos bastonetes, foram descritos mais de 150 mutações responsáveis pela forma AD(22).
Doenças com herança AR ou ligada ao X recessiva acontecem por falta de uma função de um gene. Nesse caso, a terapia gênica atua na introdução de uma cópia do gene ausente. Na herança AD, mutações em um gene específico são responsáveis pela doença. A terapia gênica visa inativar o efeito desse gene defeituoso seguido de sua reposição após correção da estrutura mutada(1,23).
Nesse contexto, para evitar que a mutação heterogênea presente no gene da rodopsina cause retinose pigmentar AD, a estratégia de terapia gênica, que está em estudo, envolve a supressão do gene RHO utilizando um RNA de interferência (RNAi) e a reposição do gene modificado de forma a escapar da supressão(24).
Essa técnica consiste na supressão dos alelos mutados. A supressão ocorre por uma sequência de nucleotídeo complementar à sequência que sofreu mutação e que está causando a doença. Após a supressão da mutação e dos alelos é realizada a reposição desse gene com o códon modificado, permitindo a correta tradução da proteína(24-25).
Recentemente a equipe de Peter Humphries obteve sucesso com esta técnica, onde realizaram pela primeira vez um experimento in vivo, podendo mostrar a eficiência desse método(25).
Através de injeção sub-retiniana de vetores AAV com o sistema de expressão do RNAi em olhos de camundongos, observaram a supressão de 88% do gene RHO. A funcionalidade do gene inserido contendo o códon modificado ficou demonstrada pelo eletrorretinografia (ERG)(25).
DOENÇA DE STARGARDT
O gene ABCA4 (8.9 kb) está localizado no braço curto do cromossomo 1 e codifica a proteína ABCR, no segmento externo dos fotorreceptores. Com herança AR, os fenótipos causados por mutações no ABCA4 incluem RP, doença de Stargardt, distrofia de cones e bastonetes e fundus flavimaculatus(26).
A proteína ABCR está envolvida no transporte ativo de all-trans-retinal e fosfatidiletanolamina pela membrana dos discos intercalares dos fotorreceptores, auxiliando a eliminação desses componentes. Perda ou redução da função da proteína ABCR causa elevação de fosfatidiletanolamina no segmento externo dos cones e no EPR, que reage com all-trans-retinal formando um composto A2E tóxico. O aumento anormal de A2E predispõe à disfunção e/ou morte do EPR seguido de perda dos fotorreceptores(27).
Nomeado de StarGen, a terapia gênica utiliza um vetor lentiviral para a transferência de um gene ABCA4 normal aos fotorreceptores(28).
Kong et al., conseguiram eliminar o acúmulo de lipofucsina após injeção sub-retiniana em camundongos com a técnica do StarGen, mostrando ser um método eficiente para o tratamento de doenças causadas por mutações no ABCA4(28).
Allocca et al., recentemente obtiveram sucesso com a alteração do vetor adeno-associado que carrega pequenos insertos de gene (4.7 kb). A alteração feita no capsídeo do vetor possibilitou a incorporação de insertos maiores que 8.9 kb, permitindo a inserção do gene ABCA4. Os resultados da pesquisa mostram significante correção dos níveis de lipofucsina (A2E), das anormalidades no EPR e na função retiniana após injeção sub-retiniana em camundongos. A atividade foi estável até seis meses, o período mais longo de tratamento descrito in vivo(29).
SÍNDROME DE USHER
A síndrome de Usher é um grupo de desordens AR caracterizadas por perda congênita neurossensorial da audição, disfunção vestibular, associada à RP.
A Usher tipo 1 (USH1) caracteriza-se por arreflexia vestibular, surdez congênita profunda, enquanto a RP aparece na infância ou juventude. Na Usher tipo 2 (USH2) as respostas vestibulares são normais, a surdez também é congênita, mas tende a ser moderada e a RP se desenvolve na juventude. A Usher tipo 3 (USH3) caracteriza-se por respostas vestibulares variadas e perda progressiva da audição e visão na adolescência(22,30).
Os sete locus para a USH1 (USH1A até G), são localizados respectivamente nos cromossomos 14q32, 11q13.5, 11p15.1, 10q21-q22, 21q21, 10q21-q22 e 17q24-q25. Os quatro locus para a USH2 (USH2A até D) foram mapeados nos cromossomos 1q41,3p24.2-p23, 5q14 e 9q32-q34. O gene da USH3 está localizado no cromossomo 3q21-q25(23). Estes genes codificam as estruturas ciliares de conexão no segmento intermediário dos fotorreceptores. Mutações nestes genes causam doenças chamadas ciliopatias, dentre elas a síndrome de Usher.
O gene USH1B localizado no cromossomo 11q13.5, primeiro a ser identificado, foi denominado MYO7A (8.1 kb). Ele pertence a uma classe chamada superfamília da miosina, molécula motora que usa energia da hidrólise do ATP para mover filamentos de actina(31).
Estudos in vivo de análise das células do epitélio pigmentado da retina de camundongos indicam que a proteína miosina VIIa (produto do gene MYO7A) transporta melanossomos e acelera a digestão de fagossomos pelas células do EPR(32).
A terapia gênica é um modo potencial de prevenção de cegueira na síndrome de Usher, já que esta é diagnosticada precocemente permitindo que o tratamento seja realizado antes que haja degeneração retiniana.
Entretanto, alguns dos genes afetados são muito grandes, dificultando seu transporte pelos vetores. Neste contexto, através de injeção sub-retiniana de vetor lentiviral, foi possível transportar o gene MYO7A até o EPR de camundongos. Para isso, utilizaram um promotor contendo elementos do promotor MYO7A nativo. Níveis de miosina VIIa foram obtidos nas células do EPR permitindo a mobilização dos melanossomos e digestão dos fagossomos na Usher 1B(33).
AMAUROSE CONGÊNITA DE LEBER
A amaurose congênita de Leber é um tipo de distrofia retiniana, com herança AR que causa cegueira infantil. As características frequentes constam de grave perda visual (ao nascimento ou entre os primeiros anos de vida) associada à RP, redução do campo visual (campo tubular), nistagmo, cegueira noturna, não-detectável ou grave redução das respostas à eletrorretinografia(22).
Atualmente, há mutações em 11 genes identificados responsáveis pela amaurose congênita de Leber: GUCY2D, RPE65, RDH12, AIPL1, C6ORF152, RPGRIP1, CRX, CRB1, CEP290, IMPDH1.
Nos fotorreceptores esses genes estão envolvidos na fototransdução, proteínas estruturais dos fotorreceptores ou com fatores de transcrição. No EPR estão associados ao metabolismo da vitamina A e ciclo visual(34-35).
Estudos recentes identificaram que RPE65 (proteína presente no EPR) é a isomerase que converte 11-cis-retinal em all-trans-retinal, sendo que, na falta dessa proteína, não há formação de pigmento visual, ocorrendo acúmulo da vitamina A por bloqueio do ciclo visual(34,36).
Nesse contexto, um grupo de pesquisadores introduziu 9-cis-retinal, via oral, em camundongos, e após 48 horas, observou a formação de fotopigmento e a rápida restauração da função retiniana pela proteína RPE65. O isômero 9-cis-retinal foi utilizado por ser mais eficiente na regeneração do pigmento visual, ter maior estabilidade e maior resistência aos ácidos estomacais. Esse estudo mostrou que a intervenção farmacológica pode ser uma ferramenta útil na prevenção da cegueira nesse tipo de distrofia(37).
Narfström et al., obtiveram sucesso ao injetar via sub-retiniana, vetores AAV carregando o gene RPE65 em olhos de cães. Observaram que a função retiniana melhorou em menos de quatro semanas, persistindo até nove meses nos animais entre dois e onze meses de vida, sendo que acima de dois anos e meio o tratamento é ineficiente já que a degeneração retiniana está em estágio mais avançado. Pela primeira vez, reportaram o aumento nas respostas da eletrorretinografia em ambos os olhos e não apenas no olho de estudo(35).
Em estudo semelhante, Acland et al., obtiveram o mesmo resultado, e puderam acrescentar que a injeção intravítrea nessas mesmas condições não é eficiente, pois só transfecta células ganglionares(36).
Recentemente, foram publicados os resultados de dois estudos em humanos. No estudo de Bainbrigde et al., dos três pacientes submetidos à injeção sub-retiniana durante vitrectomia com vetor AAV, apenas um teve melhora da acuidade visual. Os autores sugerem que a explicação está no fato da patologia estar no estágio mais brando que nos outros pacientes e a administração ser realizada no olho amblíope(38).
Maguire et al., obtiveram sucesso com estudo semelhante. Também utilizaram AAV com injeção sub-retiniana e vitrectomia no olho amblíope em três pacientes. Todos tiveram melhora da acuidade visual após seis semanas, redução do nistagmo e aumento do reflexo pupilar à luz(39).
Nos dois estudos não houve evidência de toxicidade, inflamação e efeitos adversos ao tratamento. Os autores sugerem que o tratamento poderia ser mais eficiente se realizado em crianças onde a degeneração da retina é menor podendo melhorar a função visual(38-39).
PERSPECTIVAS PARA TERAPIA GÊNICA EM DISTROFIAS RETINIANAS
A terapia gênica age na correção de um defeito no gene que causa doenças oculares e tem se mostrado eficaz no tratamento de defeitos gênicos em modelos animais.
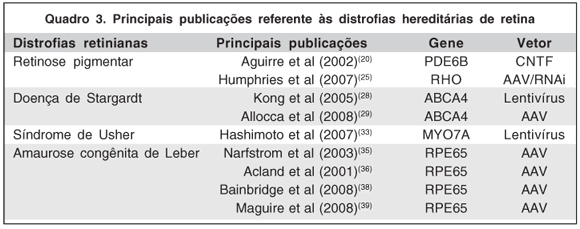
Muitas publicações contribuíram para o avanço das pesquisas nessa área (Quadro 3).
Estudos pré-clínicos começaram após décadas de estudos oftalmológicos e os resultados são promissores (Quadro 4).
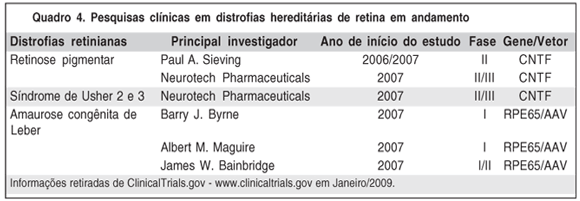
Apesar de muitos esforços ainda serem necessários para um efetivo resultado em humanos, os recentes avanços nesta área, poderão impulsionar o desenvolvimento de novas propostas para pesquisas clínicas com terapia gênica para doenças oculares hereditárias e adquiridas.
REFERÊNCIAS
1. Morales MM. Terapias avançadas: célula-tronco, terapia gênica e nanotecnologia aplicada à saúde. In: Sang WH. Introdução. História da terapia gênica, estado da arte, técnicas e ética. São Paulo: Atheneu; 2007. p.105-25.
2. Morales MM. Terapias Avançadas - Célula-tronco, terapia gênica e nanotecnologia aplicada à saúde. In: Bryan E. Strauss. Vetores para terapia gênica. Vírus, plasmídeos e outros métodos. São Paulo: Atheneu; 2007. p.126-48.
3. Ali RR, Reichel MB, Hunt DM, Bhattacharya SS. Gene therapy for inherited retinal degeneration. Br J Ophtalmol. 1997:81(9):795-801.
4. Wright AF. Gene therapy for the eye. Br J Ophthalmol. 1997:81(8):620-3.
5. Nardi NB, Teixeira LA, Silva EF. Terapia gênica. Cienc Saúde Coletiva. 2002; 7(1):109-16.
6. Lehninger AL, Nelson DL, Cox MM. Princípios de Bioquímica. São Paulo: Sarvier; 2002. p.975.
7. Takahashi M, Miyoshi H, Verma IM, Gage FH. Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J Virol. 1999;73(9):7812-6.
8. Bainbridge JW, Stephens C, Parsley K, Demaison C, Halfyard A, Thrasher AJ, et al. In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector: efficient long-term transduction of corneal endothelium and retinal pigment epithelium. Gene Ther. 2001;8(21):1665-8.
9. Li T, Adamian M, Roof DJ, Berson EL, Dryja TP, Roessler BJ, Davidson BL. In vivo transfer of a reporter gene to the retina mediated by an adenoviral vector. Invest Ophthalmol Vis Sci. 1994;35(5):2543-9.
10. Bennett J, Wilson J, Sun D, Forbes B, Maguire A. Adenovirus vector-mediated in vivo gene transfer into adult murine retina. Invest Ophthalmol Vis Sci. 1994; 35(5):253542.
11. Ali RR, Reichel MB, Thrasher AJ, Levinsky RJ, Kinnon C, Kanuga N, et al. Gene transfer into the mouse retina mediated by an adeno-associated viral vector. Human Mol Genet. 1996;5(5):591-4.
12. Jomary C, Vicent KA, Grist J, Neal MJ, Jones SE. Rescue of photoreceptor function by AAV-mediated gene transfer in a mouse model of inherited retinal degeneration. Gene Therapy. 1997;4(7):683-90.
13. Hodgson CP, Solaiman R. Virosomes: cationic liposomes enhance retroviral transduction. Nat Biotechnol. 1996;14(3):339-42.
14. Cai X., Conley S, Naash M. Nanoparticle applications in ocular gene therapy. Vision Res. 2008;48(3):319-24.
15. Farjo R, Skaggs J, Quiambao AB, Cooper MJ, Naash MI. Efficient non-viral ocular gene transfer with compacted DNA nanoparticles. PLoS ONE. 2006; Dec 20;1:e38.
16. Chong NH, Bird AC. Management of inherited outer retinal dystrophies: present and future. Br J Ophtalmol. 1999:83(1):120-2.
17. Faktorovich EG, Steinberg RH, Yasumura D, Matthes MT, LaVail MM. Photoreceptor degeneration in inherited retinal dystrophy delayed by basic fibroblast growth factor. Nature. 1990;347(6288):83-6.
18. Borhani H, Peyman GA, Rahimy MH, Beuerman RW. Vitreoretinal toxicity of basic fibroblast growth factor. Int Ophthalmol. 1993;17(4):195-9.
19. Cayouette M, Behn D, Sendtner M, Lachapelle P, Gravel C. Intraocular gene transfer of ciliary neurotrophic factor prevents death and increases responsiveness of rod photoreceptors in the retinal degeneration slow mouse. J Neurosci. 1998; 18(22):9282-93.
20. Tao W, Wen R, Goddard MB, Sherman SD, O'Rourke PJ, Stabila PF, et al. Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2002;43(10): 3292-8.
21. Gal A, Apfeistedt-Sylia E, Janecke AR, Zrennert E. Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Prog Retin Eye Res. 1997;16(1): 51-79.
22. Unonius N, Farah ME, Sallum JM. Classificação diagnóstica dos portadores de doenças degenerativas de retina, integrantes dos grupos Retina São Paulo e Retina Vale do Paraíba. Arq Bras Oftalmol. 2003;66(5):443-8.
23. Bennett J, Maguire AM. Gene therapy for ocular disease. Mol Ther. 2000; 1(6):501-6.
24. O'Reilly M, Millington-Wards S, Palfi A, Chadderton N, Cronin T, McNally N, et al. A transgenic mouse model for gene therapy of rhodopsin-linked Retinitis Pigmentosa. Vision Res. 2008;48(3):386-91.
25. O'Reilly M, Palfi A, Chadderton N, Milligton-Ward S, Ader M, Cronin T, et al. RNA interferencemediated suppression and replacement of human rhodopsin in vivo. Am J Hum Genet. 2007;81(1):127-35.
26. Weleber RG. Inherited and orphan retinal diseases: phenotypes, genotypes, and probable treatment groups. Retina. 2005;25(8 Suppl):S 4-S7.
27. Côco M, Baba NT, Sallum JM. Avaliação da autofluorescência do fundo de olho nas distrofias de retina com o aparelho Heidelberg Retina Angiograph2. Arq Bras Oftalmol. 2007;70(5):739-45.
28. Kong J, Kim SR, Binley K, Pata I, Doi K, Mannik J, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Invest Ophthalmol Vis Sci. 2005:46:E-Abstract 5210.
29. Allocca M, Doria M, Petrillo M, Colella P, Garcia-Hoyos M, Gibbs D, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008;118(5):1955-64.
30. Liarth JC, Gonçalves EA, Gonçalves JO, Neiva DM, Leal FA. Síndrome de Usher: caracteristicas clínicas. Arq Bras Oftalmol. 2002;65(4):457-61.
31. Williams DS. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vision Res. 2008;43(3):433-41.
32. Gibbs D, Kitamoto J, Williams DS. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc Natl Acad Sci USA. 2003;100(11):6481-6.
33. Hashimoto T, Gibbs D, Lillo C, Azarian SM, Legacki E, Zhang XM., et al. Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther. 2007;14(7):584-94.
34. Le Meuer G, Stieger K, Smith AJ, Weber M, Deschamps JY, Nivard D, et al. Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene Ther. 2007;14(4):292-303.
35. Narfström K, Katz ML, Ford M, Redmond TM, Rakoczy E, Bragadóttir R. In vivo gene therapy in young and adult RPE65-/- dogs produces long-term visual improvement. J Hered. 2003:94(1):317.
36. Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001;28(1):92-5.
37. Van Hooser JP, Aleman TS, He YG, Cideciyan AV, Kuksa V, Pittler SJ, et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci USA. 2000;97(15):8623-8.
38. Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358(21):2231-9. Comment in: N Engl J Med. 2008;358(21): 2282-4.
39. Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicellli J, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 2008;358(21):2240-8. Comment in: N Engl J Med. 2008;358(21):2282-4.
Endereço para correspondência:
Monique Côco
Departamento de Oftalmologia - UNIFESP
Rua Botucatu, 822
São Paulo (SP) CEP 04023-062
E-mail: [email protected]
Recebido para publicação em 15.07.2008
Última versão recebida em 16.01.2009
Aprovação em 26.01.2009
Trabalho realizado no Departamento de Oftalmologia da Universidade Federal de São Paulo - UNIFESP - São Paulo (SP) - Brasil

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.