Arq. Bras. Oftalmol. 2009; 72 (4): 10.1590/S0004-27492009000400019
Total: 2773
Ricardo Evangelista Marrocos de Aragão1; Régia Maria Gondim Ramos3; Felipe Bezerra Alves Pereira4; Andreya Ferreira Rodrigues Bezerra3; Daniel Nogueira Fernandes4
DOI: 10.1590/S0004-27492009000400019
ABSTRACT
Tay-Sachs disease is an autosomal recessive disorder of sphingolipid metabolism, caused by enzime hexosaminidase A deficiency that leads to an accumulation of GM2 in neurocytes which results in progressive loss of neurological function. The accumulation of lipid in retinal ganglion cells that leads to a chalk-white appearance of the fundus called "cherry red spot" is the hallmark of Tay-Sachs disease. It is also seen in others neurometabolic diseases as well as in central retinal artery occlusion. This case reports a child with Tay-Sachs disease in a family with four previous similar deaths without diagnostic.
Keywords: Gangliosidosis, GM2; Tay-Sachs disease; Nystagmus, pathologic; Macula lutea; Mucolipidosis; Human; Infant; Male
RESUMO
Tay-Sachs é uma doença autossômica recessiva, caracterizada pela deficiência da enzima hexosaminidase A levando ao acúmulo de esfingolipídios (GM2) em células neuronais que resulta em uma perda progressiva da função neurológica. O acúmulo de lipídios em células ganglionais da retina leva a uma aparência de mácula em cereja, característica do fundo de olho de pessoas acometidas. "Mácula em cereja" também pode ser vista em outras doenças neurometabólicas e em oclusão da artéria central da retina. Este trabalho relata o caso de um paciente com doença de Tay-Sachs em uma família com história de quatro óbitos por causas semelhantes sem diagnóstico.
Descritores: Gangliosidoses GM2; Doença de Tay-Sachs; Nistagmo patológico; Mácula lútea; Mucolipidoses; Humano; Lactente; Masculino
RELATOS DE CASOS
"Cherry red spot" in a patient with Tay-Sachs disease: case report
"Mácula em cereja" em paciente com doença de Tay-Sachs: relato de caso
Ricardo Evangelista Marrocos de AragãoI; Régia Maria Gondim RamosII; Felipe Bezerra Alves PereiraIII; Andreya Ferreira Rodrigues BezerraII; Daniel Nogueira FernandesIII
IDoutor em Medicina e Especialista em Retina e Vítreo pela Universidade de Regensburg - Alemanha. Preceptor de Retina e Vítreo da Residência Médica de Oftalmologia do Hospital Universitário Walter Cantídio da Universidade Federal do Ceará (UFC) - Fortaleza (CE) - Brasil
IIResidente do segundo ano do Serviço de Oftalmologia do Hospital Universitário Walter Cantídio da UFC - Fortaleza (CE) - Brasil
IIIResidente do terceiro ano do Serviço de Oftalmologia do Hospital Universitário Walter Cantídio da UFC - Fortaleza (CE) - Brasil
ABSTRACT
Tay-Sachs disease is an autosomal recessive disorder of sphingolipid metabolism, caused by enzime hexosaminidase A deficiency that leads to an accumulation of GM2 in neurocytes which results in progressive loss of neurological function. The accumulation of lipid in retinal ganglion cells that leads to a chalk-white appearance of the fundus called "cherry red spot" is the hallmark of Tay-Sachs disease. It is also seen in others neurometabolic diseases as well as in central retinal artery occlusion. This case reports a child with Tay-Sachs disease in a family with four previous similar deaths without diagnostic.
Keywords: Gangliosidosis, GM2; Tay-Sachs disease; Nystagmus, pathologic; Macula lutea; Mucolipidosis; Human; Infant; Male; Case reports [Publication type]
RESUMO
Tay-Sachs é uma doença autossômica recessiva, caracterizada pela deficiência da enzima hexosaminidase A levando ao acúmulo de esfingolipídios (GM2) em células neuronais que resulta em uma perda progressiva da função neurológica. O acúmulo de lipídios em células ganglionais da retina leva a uma aparência de mácula em cereja, característica do fundo de olho de pessoas acometidas. "Mácula em cereja" também pode ser vista em outras doenças neurometabólicas e em oclusão da artéria central da retina. Este trabalho relata o caso de um paciente com doença de Tay-Sachs em uma família com história de quatro óbitos por causas semelhantes sem diagnóstico.
Descritores: Gangliosidoses GM2; Doença de Tay-Sachs; Nistagmo patológico; Mácula lútea; Mucolipidoses; Humano; Lactente; Masculino; Relatos de casos [Tipo de publicação]
INTRODUCTION
Tay-Sachs disease is an autosomal recessive, progressive neurodegenerative disorder which, in the classic infatile form, is usually fatal by the age of 2 or 3 years(1). Is a sphingolipidosis characterised by deficiency of lysosomal enzymes required to sphingolipids degradation: gangliosides, cerebrosides and sphingomyelins(2). Tay-Sachs is characterized by lesions in the central nervous system due to the accumulation of GM2 trihexosylceramide in neurocytes(3), a disorder of sphingolipid metabolism caused by enzyme β hexosaminidase A deficiency(4). Infantile Tay-Sachs disease is characterized by normal development until 4 to 5 months of age, followed by progressive psychomotor retardation, megalencephaly, retinal "cherry red spot", blindness, and death by the age of 3 to 5 years(4). The term "cherry red spot" describes the retinal ophthalmoscopic appearance in neurometabolic disorders such as Tay-Sachs disease (GM2 type I) described first by Warren Tay in 1881(5) "...optic discs apparently quite healthy, but in region of the yellow spot in each eye there was a conspicuous, tolerably well-defined, large white patch, more or less circular in outline, and showing at its center a brownish-red, nearly circular spot, contrasting strongly with the white patch surrounding it."(6). This fundus appearance also accompanies other neuronal lipid-storage disorders including Sandhoff disease (GM2 type II), gangliosidosis GM2 type III and GM1 type I, Niemann-Pick disease, sialidosis types I and II, Farber disease, mucolipidosis III, metachromatic leucodystrophy, multiple sulfatase deficiency, toxicity (dapsone), and Wolman disease(7).
This fundus also resembles central retinal artery occlusion, which shows a pale retina as a result of reduced blood flow. The characteristic pale hue results from intensive deposition of lipid, sphingolipid, or oligosaccharide material in retinal ganglion cells at the macula, where cells are several layers thick. In the center of the pale region lies the foveal pit, in which lacks ganglion cells, and thus continues to retain its reddish appearance(8). The fovea pit color results from the pigment epithelium and choroids which is red only in Caucasian patients. In others races it can be a "brown spot" or a "black spot"(5).
CASE REPORT
The subject was a male aged 1 year and 7 months, who came to the university hospital brought by his mother by seizures, cough, and fever for about one week.
He had gone to the hospital twice before due to otitis and pneumonia.
His mother related 4 relatives who died before the completion of two years of life.
His parents were healthy and first cousins.
He was born at a gestational age of 36 weeks. When the baby was one month old, it was observed that the baby had little power for holding its head or moving its limbs. Since then, the weakness has become progressively evident. The mother noticed that the baby did not see as other children of the same age.
In the hospital initial exam the child had fever, rhonchi on chest auscultation and marasmus-like secondary protein-energy malnutrition. The neurological examination revealed psychomotor retardation, horizontal and bilateral nistagmus, muscle weakness, generalized hyperreflexia, clumsiness, and presence of Babinski and Moro signal. The ophthalmoscopic examination was very difficult due to the nystagmus, and showed chalk-white macular areas with a "cherry red spot" in the center of both eyes (Figure 1).
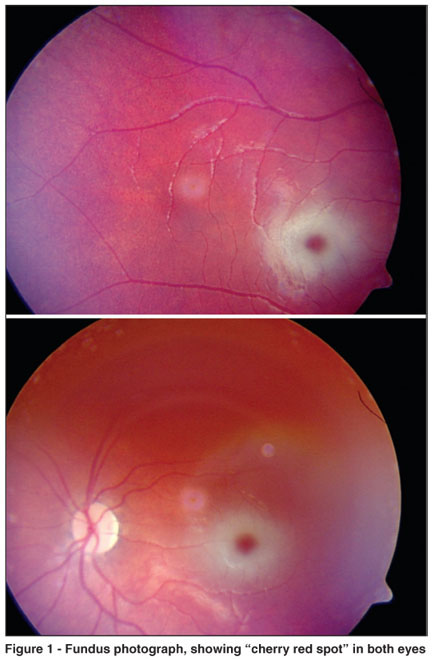
Assaying for the activity of β hexosaminidase A in the serum revealed a deficiency of this enzyme, whereby the diagnosis of Tay-Sachs disease was done.
DISCUSSION
The pathogenesis of Tay-Sachs disease is attributable to the accumulation of GM2 trihexosylceramide secondary to defects of β hexosaminidase A enzyme, caused by mutation in the alpha subunit of the hexominidase A gene on chromosome 15q(1). GM2 trihexosylceramide accumulates predominantly in retinal ganglion cells whereby retina becomes turbid with milky-white coloration(3). Gangliosidosis is most plentiful in the gray matter with the most of clinical and pathologic manifestations on the nervous system(8). This is typically present early in the course of illness, is frequently a helpful clue in diagnosis and has been detected even in the fetal stage (4). As the ganglion cells atrophy and disappear, the cherry red spot may become less prominent and blindness with optic atrophy eventually ensues. Infantile Tay-Sachs disease is also characterized by progressive loss of neurological function, hyperirritability, and progressive weakness. The infants rapidly lose motors and intellectual skills after the end of the first year and may crawl but never walk. The natural course leads to a decerebrate, vegetative state by second to fourth year of life and death(8). A juvenile form of Tay-Sachs disease exists, with late onset and slower course but a similar constellation of signs. The mildest form of the disease is the adult sub-type, also called late-onset Tay-Sachs disease (LOTS). Manifestations include ataxia, dysarthria, muscle weakness, and dementia. But, in that form, no cherry red spot is seen for the reason explained above(4). One final variant is the chronic form, with patients surviving well into adulthood. There are approximately 78 described mutations although the majority of patients have the infantile form(4).
The diagnosis of the GM2 gangliosidosis is accomplished by assaying for the activity of individual β hexosaminidase isoenzymes in serum or cultured cells from affected individuals(8).
Enzyme replacement therapies (ERT) have been introduced for several lysosomal storage, and intensive studies are being carried out to develop others therapies, but the treatment of the neurological symptoms of these diseases is still not pos-sible(9-10). Small molecule therapy called pharmacological chaperones (PC) that can stabilize the conformation of a mutant protein and also acts as competitive inhibitors(11) has been shown to successfully enhance the enzymes level in Tay-Sachs disease(12). Substrate reduction therapy that uses small molecules to slow the rate of glycolipid biosynthesis shows efficacy in mouse models of Tay-Sachs(13). The therapy called the stop codon read-through strategy is based on the use of specific chemicals, which cause the misuse of stop codons during translation on ribosomes, and incorporation of an amino acid instead of translation termination. This may have a therapeutic effect in patients lacking a particular gene product. Such strategy is still at the stage of laboratory experiments(9). Gene therapy has the potential for widespread correction of the underlying lysosomal defect by means of the secretion-recapture cellular pathway for enzymatic complementation. Gene delivery of β hexosaminidase A by using adeno-associated viral vectors has realistic potential for treating the human Tay-Sachs-related diseases(14).
With more effective means of treatment, several strategies may become available for improving lives of patients suffering from neuronopathic forms of inherited lysosomal storage diseases.
REFERENCES
1. OMIM - Online Mendelian Inheritance in Man. Tay-Sachs Disease. 272800. [Internet]. Baltimore MD: National Center for Biotechnology Information [cited 2008 Jan 12]. Available from:. http://www.ncbi.nlm.nih.gov/entrez/dispomim. cgi?id=272800
2. Sallum JM. Genética ocular. In: Conselho Brasileiro de Oftalmologia. Bases da Oftalmologia. São Paulo: Cultura Médica; 2008. v.1, p.461-99. (Série Brasileira de Oftalmologia)
3. Nakata-Onishi M, Suzuki A, Okamoto N, Fukada M. Observations on time course changes of the cherry red spot in a patient with Tay-Sachs disease. Br J Ophthalmol. 2000;84(1):1320-1.
4. Rucker JC, Shafiro BE, Han YH, Kumar AN, Garbutt S, Keller EL, et al. Neuro-ophthalmology of late-onset Tay-Sachs disease (LOTS). Neurology. 2004; 63(10):1918-26.
5. Ospina LH, Lyons CJ, McCormick AQ. "Cherry-red spot" or "perifoveal white patch"? Can J Ophthalmology. 2005;40(5):609-10.
6. Koller C. A rare and fatal disease of infancy. Trans Am Ophthalmol Soc. 1896; 7:661-8.
7. Leavitt JA, Kotagal S. The "cherry red" spot. Pediatr Neurol. 2007;37(1):74-5.
8. Palmer JD, Mukai S. Metabolic diseases. In: Regillo CD, Brown GC, Flynn Jr. HW, editors. Vitreoretinal disease. New York: Thieme; 1999. p.347-64.
9. Jakobkiewicz-Banecka J, Wegrzyn A, Wegrzyn G. Substrate deprivation therapy: a new hope for patients suffering from neuronopathic forms of inherited lysosomal storage diseases. J Appl Genet. 2007;48(4):383-8.
10. Akeboshi H, Chiba Y, Kasahara Y, Takashiba M, Takaoka Y, Ohsawa M, et al. Prodution of recombinant beta-hexosaminidase A, a potential enzyme for replacement therapy for Tay-Sachs and Sandhoff diseases, in the methylotrophic yeast Ogataea minuta. Appl Environ Microbiolol. 2007;73(15):4805-12.
11. Maegawa GH, Tropak M, Buttner J, Stockley T, Kok F, Clarke JT, et al. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J Biol Chem. 2007;282(12):9150-61.
12. Tropak MB, Blanchard JE, Withers SG, Brown ED, Mahuran D. High-throughput screening for human lysosomal beta-N-Acetyl hexosaminidase inhibitors acting as pharmacological chaperones. Chem Biol. 2007;14(2):153-64. Comment in: Chem Biol. 2007;14(3):241-2.
13. Platt FM, Jeyakumar M, Andersson U, Heare T, Dwek RA, Butters TD. Substrate reduction therapy in mouse models of the glycosphingolipidoses. Philos Trans R Soc Lond B Biol Sci. 2003;358(1433):947-54.
14. Cachón-Gonzalez MB, Wang SZ, Lynch A, Ziegler R, Cheng SH, Cox TM. Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proc Natl Acad Sci USA. 2006;103(27):10373-8.
 Endereço para correspondência:
Endereço para correspondência:
Ricardo E. Marrocos de Aragão
Rua Osvaldo Cruz, 2.335
Fortaleza (CE) CEP 60125-151
E-mail: [email protected]
Recebido para publicação em 20.07.2008
Última versão recebida em 23.02.2009
Aprovação em 08.04.2009
Trabalho realizado no Serviço de Oftalmologia do Hospital Universitário Walter Cantídio da Universidade Federal do Ceará (UFC) - Fortaleza (CE) - Brasil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Juliana Maria Ferraz Sallum sobre a divulgação de seu nome como revisora, agradecemos sua participação neste processo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.