Arq. Bras. Oftalmol. 2009; 72 (3): 10.1590/S0004-27492009000300028
Total: 1838
Francisco Max Damico1; Felipe Theodoro Bezerra1; Gaspar Carvalho da Silva3; Fábio Gasparin4; Joyce Hisae Yamamoto5
DOI: 10.1590/S0004-27492009000300028
ABSTRACT
Vogt-Koyanagi-Harada disease (VKH), a well-established multiorgan disorder affecting pigmented structures, is an autoimmune disorder of melanocyte proteins in genetically susceptible individuals. Several clinical and experimental data point to the importance of the effector role of CD4+ T cells and Th1 cytokines, the relevance of searching a target protein in the melanocyte, and the relevance of the HLA-DRB1*0405 in the pathogenesis of the disease. Vogt-Koyanagi-Harada disease has a benign course when early diagnosed and adequatey treated. Full-blown recurrences are rare after the acute stage of Vogt-Koyanagi-Harada disease is over. On the other hand, clinical findings, such as progressive tissue depigmentation (including sunset glow fundus) and uveitis recurrence, indicate that ocular inflammation may persist after the acute phase. Additionally, indocyanine green angiography findings suggest the presence of choroidal inflammation in eyes without clinically detectable inflammation. The aim of this paper is to review the latest research results on Vogt-Koyanagi-Harada disease pathogenesis and chronic/convalescent stages, which may help to better understand this potentially blinding disease and to improve its treatment.
Keywords: Uveomeningoencephalitic syndrome; Uveitis; Chronic disease; Autoimune diseases; HLA-DR antigens; Melanocytes; Review
RESUMO
A doença de Vogt-Koyanagi-Harada (VKH) afeta vários órgãos que têm em comum a presença de pigmento. É doença autoimune que agride os melanócitos de indivíduos geneticamente susceptíveis. Inúmeras evidências clínicas e experimentais demonstram a importância de células T CD4+ como células efetoras da resposta imune celular, das citocinas pró-inflamatórias Th1, da procura da proteína-alvo dentro do melanócito, e da relevância do HLA-classe II DRB1*0405 na patogênese desta doença. A doença de Vogt-Koyanagi-Harada apresenta bom prognóstico visual desde que o diagnóstico seja precoce e o tratamento instituído seja adequado. Recidivas com acometimento do segmento posterior são raras após a fase aguda da doença. No entanto, achados clínicos como a progressiva despigmentação do fundo, incluindo o aspecto em por do sol, e as recidivas da uveíte indicam que a inflamação ocular pode persistir mesmo após a fase aguda da doença. Os achados da angiografia com indocianina verde também sugerem a presença de inflamação da coróide mesmo em olhos sem inflamação clinicamente detectável. O objetivo do presente trabalho é rever os mais recentes estudos sobre a patogênese da doença Vogt-Koyanagi-Harada e sobre os aspectos clínicos da fase crônica e/ou convalescente da doença, permitindo melhores conhecimentos sobre esta doença potencialmente mórbida e oferecendo terapias mais adequadas.
Descritores: Síndrome uveomeningoencefálica; Uveíte; Doença crônica; Doenças autoimunes; Antígenos HLA-DR; Melanócitos; Revisão
CURRENT UPDATE
New insights into Vogt-Koyanagi-Harada disease
Novos conhecimentos sobre a doença de Vogt-Koyanagi-Harada
Francisco Max DamicoI; Felipe Theodoro BezerraI; Gaspar Carvalho da SilvaII; Fábio GasparinIII; Joyce Hisae YamamotoIV
IAttending Physician, Hospital das Clínicas, University of São Paulo Medical School. Researcher, Neuroscience and Behavior, Department of Experimental Psychology, Universidade de São Paulo - USP - São Paulo (SP) Brazil
IIPost-Graduation Student, Department of Ophthalmology, USP - São Paulo (SP) - Brazil
IIIFormer Chief Resident, Hospital das Clínicas, Post-Graduation Candidate, USP - São Paulo (SP) - Brazil
IVSenior Researcher, Uveitis Service, Hospital das Clínicas, USP - São Paulo (SP) - Brazil
ABSTRACT
Vogt-Koyanagi-Harada disease (VKH), a well-established multiorgan disorder affecting pigmented structures, is an autoimmune disorder of melanocyte proteins in genetically susceptible individuals. Several clinical and experimental data point to the importance of the effector role of CD4+ T cells and Th1 cytokines, the relevance of searching a target protein in the melanocyte, and the relevance of the HLA-DRB1*0405 in the pathogenesis of the disease. Vogt-Koyanagi-Harada disease has a benign course when early diagnosed and adequatey treated. Full-blown recurrences are rare after the acute stage of Vogt-Koyanagi-Harada disease is over. On the other hand, clinical findings, such as progressive tissue depigmentation (including sunset glow fundus) and uveitis recurrence, indicate that ocular inflammation may persist after the acute phase. Additionally, indocyanine green angiography findings suggest the presence of choroidal inflammation in eyes without clinically detectable inflammation. The aim of this paper is to review the latest research results on Vogt-Koyanagi-Harada disease pathogenesis and chronic/convalescent stages, which may help to better understand this potentially blinding disease and to improve its treatment.
Keywords: Uveomeningoencephalitic syndrome; Uveitis/etiology; Chronic disease; Autoimune diseases/immunology; HLA-DR antigens; Melanocytes/immunology; Review
RESUMO
A doença de Vogt-Koyanagi-Harada (VKH) afeta vários órgãos que têm em comum a presença de pigmento. É doença autoimune que agride os melanócitos de indivíduos geneticamente susceptíveis. Inúmeras evidências clínicas e experimentais demonstram a importância de células T CD4+ como células efetoras da resposta imune celular, das citocinas pró-inflamatórias Th1, da procura da proteína-alvo dentro do melanócito, e da relevância do HLA-classe II DRB1*0405 na patogênese desta doença. A doença de Vogt-Koyanagi-Harada apresenta bom prognóstico visual desde que o diagnóstico seja precoce e o tratamento instituído seja adequado. Recidivas com acometimento do segmento posterior são raras após a fase aguda da doença. No entanto, achados clínicos como a progressiva despigmentação do fundo, incluindo o aspecto em por do sol, e as recidivas da uveíte indicam que a inflamação ocular pode persistir mesmo após a fase aguda da doença. Os achados da angiografia com indocianina verde também sugerem a presença de inflamação da coróide mesmo em olhos sem inflamação clinicamente detectável. O objetivo do presente trabalho é rever os mais recentes estudos sobre a patogênese da doença Vogt-Koyanagi-Harada e sobre os aspectos clínicos da fase crônica e/ou convalescente da doença, permitindo melhores conhecimentos sobre esta doença potencialmente mórbida e oferecendo terapias mais adequadas.
Descritores: Síndrome uveomeningoencefálica; Uveíte/etiologia; Doença crônica; Doenças autoimunes/imunologia; Antígenos HLA-DR; Melanócitos/imunologia; Revisão
INTRODUCTION
Vogt-Koyanagi-Harada disease (VKH) is a well-established multiorgan disorder that affects pigmented structures, such as the eye, inner ear, meninges, and skin(1). It is an inflammatory condition of autoimmune nature in which cytotoxic T cell target melanocytes in genetically susceptible individuals. VKH is one of the most common causes of uveitis in Japan and Brazil. The classic clinical course of VKH can be divided into four stages(1). The prodromal stage, preceding the acute uveitic stage by a few days, may mimic a viral infection. The acute uveitic stage, that lasts several weeks, is followed by the convalescent stage, in which the depigmentation of the tissues is more evident. Some patients go into the chronic recurrent stage with recurrent bouts of anterior uveitis.
The criteria proposed in the past have been progressively substituted by the International Nomenclature Committee Revised Diagnostic Criteria. The Revised Diagnostic Criteria (RDC) are more comprehensive as patients in both acute and chronic stages are contemplated and some of the ancillary examinations are considered in detail(2). It classifies patients into 3 categories (complete, incomplete, and probable VKH disease) according to the presence of extraocular manifestations. Although the importance of these different disease categories has not been shown in retrospective studies, prospective studies should be undertaken as extraocular manifestations may be short-lasting, possibly influenced by treatment. Imaging technologies, such as indocyanine green angiography (ICGA) and optical coherence tomography, have proven very useful in the diagnosis and follow-up of VKH patients and may be contemplated in future revisions of the RDC. Knowing the RDC and applying them early in the course of acute VKH is mandatory, as evidences point out that early and vigorous treatment may influence outcome. Also of importance is recognizing and diagnosing chronic phase patients, which may otherwise be treated as other uveitic entities. It seems improbable that patients with VKH disease may go unnoticed once correctly evaluated in either phase(3-4).
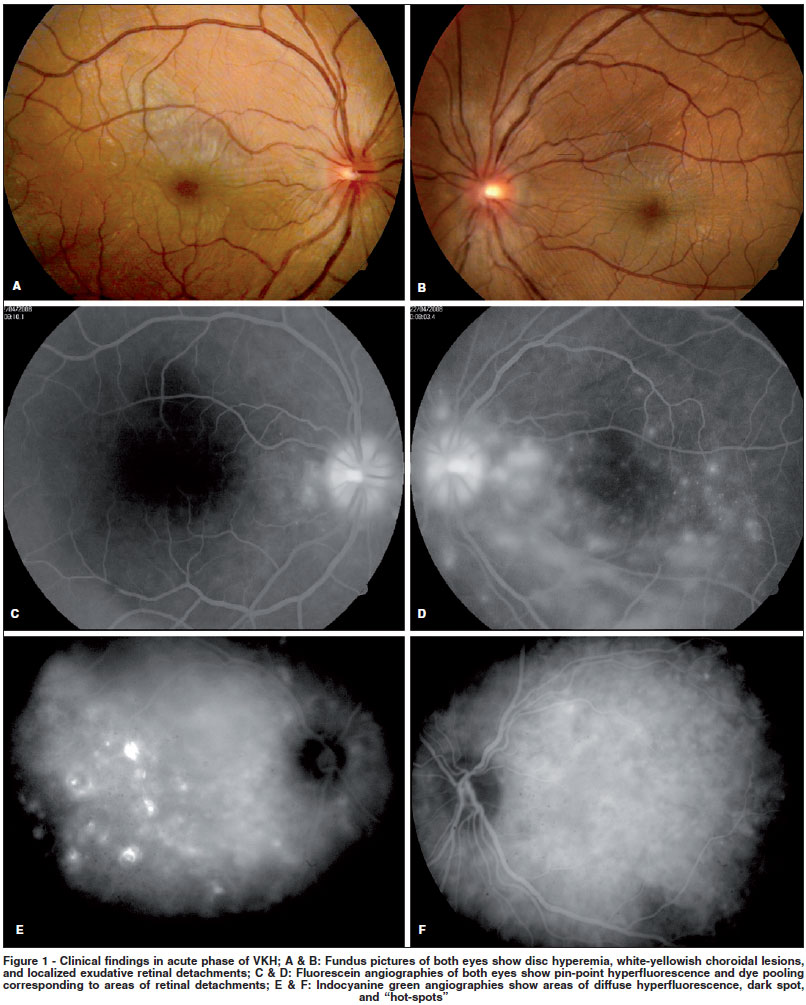
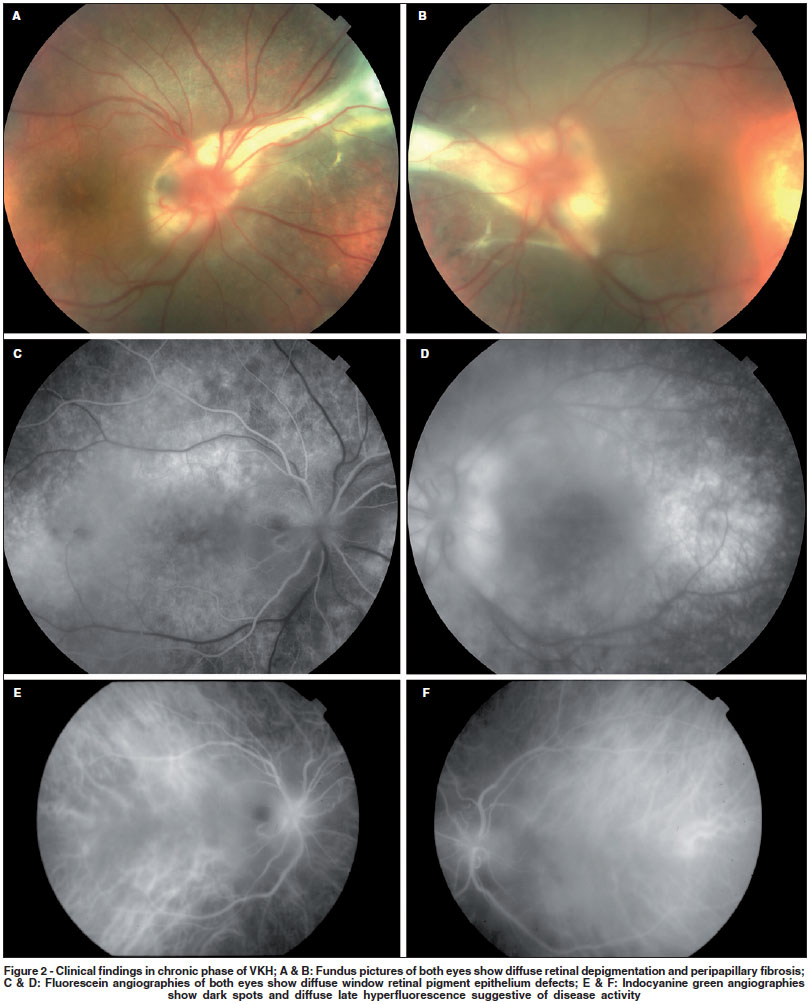
Although diagnosis of acute VKH is generally straightforward (Figure 1) and treatment tends to be effective in most patients, little is known about convalescent and chronic stages. Full-blown recurrences are rare after the acute stage of VKH is over. On the other hand, clinical findings, such as progressive tissue depigmentation (including sunset glow fundus) and uveitis recurrence indicate that systemic and ocular inflammation may persist after the acute phase(5-6)(Figure 2). Additionally, ICGA findings suggest the presence of choroidal inflammation in eyes without clinically detectable inflammation(7).
The aim of this paper is to review the latest research results on VKH pathogenesis and chronic/convalescent stages, which may help to better understand this potentially blinding disease and to improve its treatment.
AUTOIMMUNITY AGAINST MELANOCYTES
Clinical features of choroidal and skin depigmentation were the first evidence of melanocyte role in the pathogenesis, and histopathology findings support it. Transmission electron microscopy of eyes in the early stage shows a close contact between melanocytes and lymphocytes in the uvea. Histopathology of eyes in the end stage of VKH shows a remarkable disappearance of choroidal melanocytes, and immunohistochemistry identified T and B lymphocytes in the choroids(1).
Role of CD4+ T cells
Functional in vitro assays further contribute to the understanding of the effector cell in this autoimmune response against pigmented cells. Uveal pigment inhibits leukocyte migration of peripheral blood mononuclear cells (PBMC) of patients with VKH. Cytotoxic leukocytes against melanoma cells are present in peripheral blood and cerebral spinal fluid (CSF) of patients with VKH. Cytotoxic CD4+ and CD8+ T lymphocytes against human melanoma cells and melanocytes are present in the peripheral blood of patients with VKH(8). These findings support the main role of CD4+ T cells in the pathogenesis of VKH.
Immunohistochemical studies show that eyes with active VKH present activated T lymphocytes in the choroid which express both early and late activation markers. Activated CD4+ T cells found in the skin in depigmented area(9) and melanin-laden macrophages described in CSF of patients with VKH reinforce the role of the melanocyte and systemic nature of the disease.
Cytokines are major regulators of leukocyte traffic under physiological and pathological conditions. Th1 cytokine profile is a common finding in most human organ-specific autoimmune diseases. Th1 cytokine profile is described in PBMC of patients with VKH. T cell clones from infiltrating cells of the aqueous humor and PBMC of patients spontaneously produce larger amounts of IL-6, IL-8, and IFN-γ(10). Gocho et al. isolated T cell clones from PBMC of patients specific for tyrosinase family proteins with predominant Th1 cytokine profile(11). Damico et al. demonstrated that PBMC of patients recognize an increased breadth of melanocyte protein-derived peptides with higher affinity than control subjects and do not produce IL-4, a Th2 cytokine, in response to disease-specific peptides(12). Differences in cytokine concentrations in CSF and sera of patients with VKH corroborate a Th1-dominant condition in VKH(13).
Recently, two new T helper subsets were described in mice and humans: T helper 17 (Th17) and regulatory T cells (Treg). Th17 may be important in the development and progression of inflammatory and autoimmune diseases and Tregs are characterized by their immunoregulatory ability to inhibit the development of certain autoimmune diseases in animal models. Both T helper subsets were described in VKH(14-15).
Immunogenetics
Genetic factor may play an important role in tolerance breakdown in VKH. The strong association of VKH with HLA-DR4/ DR53 in the Japanese population has long been known(16). This association was also found in Chinese, North American, and Hispanic patients(17). A secondary association with HLA-DR1 was also described, involving a shared sequence linked to susceptibility to rheumatoid arthritis. Goldberg et al. showed that HLA-DRB1*0405 is also the predominant susceptibility allele in Brazilian patients, a very ethnic heterogeneous population. In that study, the different alleles originally proposed to be associated with VKH were present separately, reinforcing the importance of HLA-DRB1*0405 as the main susceptibility allele to VKH(18).
The importance of this allele in susceptibility to the development of VKH was reinforced by Damico et al., who showed that patients bearing HLA-DRB1*0405 recognized a larger melanocyte-derived peptide repertoire than HLA-matched control subjects(12).
Search for a target in VKH
Melanocyte differentiation proteins are the most studied proteins in VKH pathogenesis. These proteins, tyrosinase, tyrosinase-related protein 1 (TRP-1), tyrosinase-related protein 2 (TRP-2), gp100/PMel-17, and MART-1/Melan A, were first described in melanoma-related studies and are recognized by T cells from patients with melanoma.
Melanocyte proteins, believed to be expressed in melanosomes, are involved in the synthesis of melanin pigment. Most of those proteins were identified as melanoma antigens by screening cDNA library with HLA-class I-restricted melanoma-reactive cytotoxic T lymphocytes. In pigmented cells, melanin is synthesized within melanosomes through enzymatic reactions from tyrosine. The first reactions are catalyzed by tyrosinase which is regarded as the key enzyme in melanin synthesis. Tyrosinase and the two other related enzymes, TRP-1 and TRP-2, play an important role in the initiation of the process of melanization, and are expressed in the eye, skin, and brain.
Tyrosinase, TRP-1 and TRP-2. Several authors showed cellular immune response of peripheral T cells or T cells from intraocular fluid against peptides derived from tyrosinase and its related proteins(12,19).
MART-1. Sugita et al. isolated CD8+ T cell clones from intraocular fluid with a specific cytotoxicity activity against MART-1 in HLA-A2-positive patients with VKH(20).
PMel-17/gp100. Chan et al. showed that experimental melanin-protein induced uveitis can be ameliorated by a recombinant adenovirus construct encoding gp100(21). However, some authors studied peptides derived from this protein but VKH relevant peptides were not found in VKH patients(12).
Several peptides selected from tyrosinase, TRP-1 and TRP2 are restricted to HLA-DRB1*0405 and are potential candidates for the autoimmune response observed in VKH. Evidences from experimental VKH, established by Yamaki et al., with tyrosinase, TRP-1, and TRP-2 peptides reinforce the role of these proteins in VKH pathogenesis(22). In spite of these studies, the exact role of tyrosinase and its related proteins in the pathogenesis of VKH has not yet been clarified. Other attempts to identify additional autoantigens involved in the antimelanocyte immune responses in VKH are under investigation.
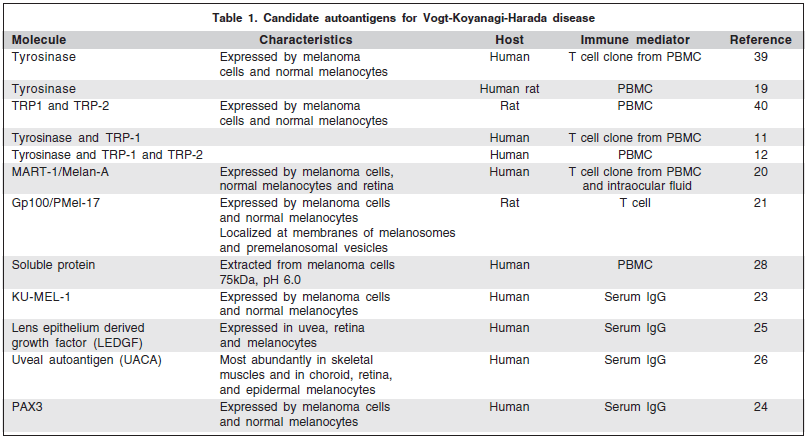
Serological identification of tumor antigens by cDNA expression cloning with sera from a melanoma patient who developed anti-melanocyte/melanoma immune responses identified an antigen called KU-MEL-1. KU-MEL-1 is an antigen strongly expressed in most melanoma cell lines, melanoma tissue samples, and cultured melanocytes but weakly expressed in cell lines derived from other types of tumors. Antibody anti-KUMEL-1 was detected in patients with VKH and was related to some clinical aspects of the disease(23). A protein called PAX3 and its isoform were isolated from melanoma and melanocytes at relatively high frequency by systematic gene expression analysis combined with rapid screening of patient sera for immunogenicity against a highly pigmented melanoma cell line. Antibodies against this protein were detected in some patients with melanoma and VKH, but not in healthy subjects(24). Two other autoantigens - lens epithelium derived growth factor (LEDGF) and uveal autoantigen (UACA) - were also isolated by cDNA cloning with a cDNA library made from bovine uvea and sera from patients with VKH(25-26).
Further methods for prediction of binding of putative antigenic peptides to HLA-DRB1 alleles are recently being proposed(27).
Table 1 summarizes the candidate autoantigens in VKH.
Infection and autoimmunity in VKH
Concerning the etiology of autoimmune diseases, efforts are focused on the search for triggering factors(28). Viral infections and autoimmune disease have long been linked. Viral symptoms often precede inflammatory signs in the target organ. Mechanisms used to explain the association of autoimmunity and viral infection include molecular mimicry, bystander activation, and viral persistence. VKH classically starts with vague systemic symptoms suggestive of viral infection but a clear association between a specific viral agent and the disease has not been established yet. Molecular studies utilizing polymerase chain reaction showed presence of Epstein-Barr virus genome in the CSF and vitreous of patients with VKH(29). Some authors described a cross-reactive T cell response between tyrosinase peptides and cytomegalovirus antigen in patients with VKH(30). Those inconclusive data point to the need for further studies to clarify the trigger of the autoimmune reaction in VKH.
OUTER RETINA AUTOIMMUNITY AND VKH
Cell-mediated and humoral immunity to photoreceptor antigens were demonstrated in VKH(31-32). However, retinal autoimmunity is also found in other types of uveitis as well as in non-inflammatory retinal disorders, such as diabetic retinopathy, particularly after argon laser photocoagulation. Therefore, the precise pathogenic role of retinal autoimmunity in VKH is still uncertain. It may simply represent an epiphenomenon that develops after retinal damage and may perpetuate inflammation that was initiated by melanocyte specific immune activation.
APOPTOSIS IN VKH
Apoptosis plays a central role in regulating the development of lymphocytes and in their homeostasis. A breakdown in apoptosis-related signaling mechanisms could result in the development of autoimmune disorders. During the past decade, a lot has been known about the various apoptotic signaling pathways and their aberrant behavior in autoimmune disorders.
Specifically in VKH, lymphocytes of patients with the disease were shown to be relatively resistant to apoptosis mediated by anti-Fas antibody(33). In addition, the strong Bcl-2 expression by CD4+ T lymphocytes of the CSF of patients with VKH, may regulate CNS apoptosis of inflammatory cells(34).
CHRONIC AND CONVALESCENT STAGES
Studies including patients with chronic and convalescent VKH have contributed to a better understanding of the clinical features of the disease.
Immunohistochemical studies showing lymphocytes in the choroid of eyes in convalescent stage suggest the presence of active ocular inflammation after acute stage manifestations. Sunset glow fundus, a very characteristic finding in convalescent and chronic stages, seems to worsen over time more rapidly in eyes with inflammation than in eyes without manifest inflammation(5). Chronic inflammation has been reported in 17.5% of patients treated with high-dose corticosteroid therapy from initial onset(6). In those 80 patients, sunset glow-fundus had a prevalence of 62% in patients with VKH without chronic ocular inflammation versus 93% in those with chronic inflammation(6). Additionally, some patients do not present sunset glow fundus after high-dose corticosteroid therapy, suggesting that this finding may be related to poor inflammatory control. Functional tests support the above findings. Multifocal electroretinogram responses are reduced in patients in convalescent stage with no apparent retinal atrophy and normal visual acuity, also indicating subclinical retinal damage(35).
ICGA is a useful method of assessing choroidal status. Typical signs of choroiditis were described in eyes with VKH in acute stage(7,36). Recently, ICGA of eyes in chronic stage with apparent isolated anterior recurrence disclosed early choroidal stromal vessel hyperfluorescence and leakage, suggesting subclinical choroiditis(37). Authors also reported two contralateral eyes without any sign of inflammation which presented the same ICGA findings(37). The most frequent ICGA finding in eyes with subclinical choroidal inflammation is the presence of hypofluorescent dark dots in intermediate phase that become isofluorescent in the late phase (Figure 2). It is hypothesized that they represent choroidal granulomas. Other less common findings may be present, such as fuzzy vascular pattern of large stromal vessels, optic disc hyperfluorescence, and choriocapillaris filling delay. There are evidences that ICGA is very useful to monitor the response to corticosteroid therapy by allowing semi-quantification of the degree of choroidal inflammation(7). Interestingly, ICGA signs of active choroidal inflammation during chronic or convalescent stages show significant decrease after systemic corticosteroid therapy, confirming the inflammatory nature of the findings(37).
CONCLUSIONS
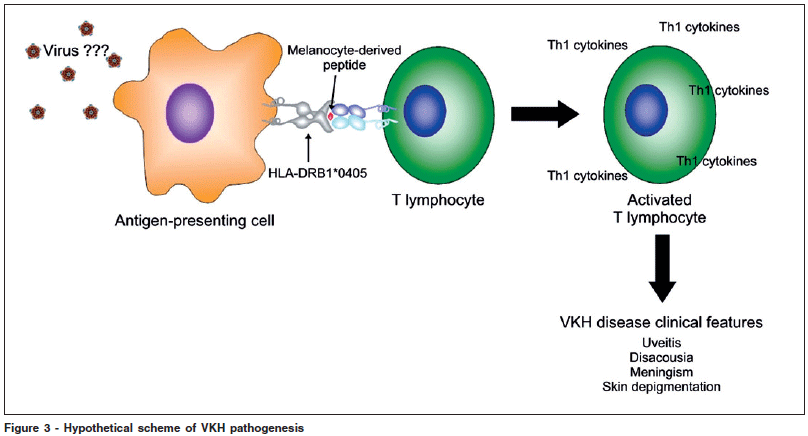
Although the clinical picture of VKH is well established, little is known about its pathogenesis. Based on the current evidences, our simplified hypothesis for the pathogenesis of VKH is summarized in Figure 3. Essentially, CD4+ T cells reactive to melanocyte specific proteins are triggered by an infectious agent. The presence of HLA-DRB1*0405 facilitates this process. This pathological condition may unmask cryptic or subdominant self-epitopes from melanocyte specific proteins and activate T cells that are normally silent(38). In perspective, questions regarding the relevance of HLA-DRB1*0405, the nature of the effector T cells, the trigger and the relevant self-antigens in the pathogenesis of VKH should be further analyzed by future research.
Clinically, there is evidence supporting the concept of ongoing choroidal inflammation even after high-dose corticosteroid therapy and apparent quiescence in the eyes. As such, initial therapy may not be sufficient to completely eradicate choroidal inflammation in all patients. ICGA seems to be a sensitive method to assess choroidal involvement and an important tool during the follow-up period. Further studies are needed to better understand the clinical importance of ongoing choroidal inflammation, its effect on retinal function and the impact its presence or eradication may have on vision in the long term.
REFERENCES
1. Moorthy RS, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol. 1995:39(4):265-92.
2. Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report an international comittee on nomenclature. Am J Ophthalmol. 2001;131(5):647-52.
3. da Silva FT, Damico FM, Marin ML, Goldberg AC, Hirata CE, Takiuti PH, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: considerations on the different disease categories. Am J Ophthalmol. 2009;147(2):339-45.
4. Cardoso IH, Zajdenweber ME, Muccioli C, Fimamor LP, Belfort R Jr. Applicability of the 2001 revised diagnostic criteria in Brazilian Vogt-Koyanagi-Harada disease patients. Arq Bras Oftalmol. 2008;71(1):67-70.
5. Suzuki S. Quantitative evaluation of "sunset glow" fundus in Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol. 1999;43(4):327-33.
6. Keino H, Goto H, Usui M. Sunset glow fundus in Vogt-Koyanagi-Harada disease with or without chronic ocular inflammation. Graefes Arch Clin Exp Ophthalmol. 2002;240(10):878-82.
7. Herbort CP, Mantovani A, Bouchenaki N. Indocyanine green angiography in Vogt-Koyanagi-Harada disease: angiographic signs and utility in patient follow-up. Int Ophthalmol. 2007;27(2-3):173-82.
8. Maezawa N, Yano A, Taniguchi M, Kojima S. The role of cytotoxic T lymphocytes in the pathogenesis of Vogt-Koyanagi-Harada disease. Ophthalmologica. 1982;185(3):179-86.
9. Okada T, Sakamoto T, Ishibashi T, Inomata H. Vitiligo in Vogt-Koyanagi-Harada disease: immunohistological analysis of inflammatory site. Graefes Arch Clin Exp Ophthalmol. 1996;234(6):359-63.
10. Sakaguchi M, Sugita S, Sagawa K, Itoh K, Mochizuki M. Cytokine production by T cells infiltrating in the eye of uveitis patients. Jpn J Ophthalmol. 1998;42(4):262-8.
11. Gocho K, Kondo I, Yamaki K. Identification of autoreactive T cells in Vogt-Koyanagi-Harada disease. Invest Ophthalmol Vis Sci. 2001;42(9):2004-9.
12. Damico FM, Cunha-Neto E, Goldberg AC, Iwai LK, Marin ML, Hammer J, et al. T-cell recognition and cytokine profile induced by melanocyte epitopes in patients with HLA-DRB1*0405-positive and -negative Vogt-Koyanagi-Harada uveitis. Invest Ophthalmol Vis Sci. 2005;46(7):2465-71.
13. Miyazawa I, Abe T, Narikawa K, Feng J, Misu T, Nakashima I, et al. Chemokine profile in the cerebrospinal fluid and serum of Vogt-Koyanagi-Harada disease. J Neuroimmunol. 2005;158(1-2):240-4.
14. Chi W, Yang P, Li B, Wu C, Jin H, Zhu X, et al. IL-23 promotes CD4+ T cells to produce IL-17 in Vogt-Koyanagi-Harada disease. J Allergy Clin Immunol. 2007;119(5):1218-24.
15. Chen L, Yang P, Zhou H, He H, Ren X, Chi W, et al. Diminished frequency and function of CD4+CD25 high regulatory T cells associated with active uveitis in Vogt-Koyanagi-Harada syndrome. Invest Ophthalmol Vis Sci. 2008; 49(8):3475-82.
16. Tagawa Y, Sugiura S, Yakura H, Wakisaka A, Aizawa M. Letter: HLA and Vogt-Koyanagi-Harada syndrome. N Engl J Med. 1976;295(3):173.
17. Davis JL, Mittal KK, Freidlin V, Mellow SR, Optican DC, Palestine G, et al. HLA associations and ancestry in Vogt-Koyanagi-Harada disease and sympathetic ophthalmia. Ophthalmology. 1990;97(9):1137-42.
18. Goldberg AC, Yamamoto JH, Chiarella JM, Marin ML, Sibinelli M, Neufeld R, et al. HLA-DRB1*0405 is the predominant allele in Brazilian patients with Vogt-Koyanagi-Harada disease. Hum Immunol. 1998;59(3):183-8.
19. Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinase family proteins are antigens specific to Vogt-Koyanagi-Harada disease. J Immunol. 2000;165(12):7323-9.
20. Sugita S, Sagawa K, Mochizuki M, Shichijo S, Itoh K. Melanocyte lysis by cytotoxic T lymphocytes recognizing the MART-1 melanoma antigen in HLA-A2 patients with Vogt-Koyanagi-Harada disease. Int Immunol. 1996;8(5):799-803.
21. Chan CC, Ying L, Sun B, Li Q, Matteson DM, Shen DF, et al. Recombinant adenovirus encoding gp100 modulates experimental melanin-induced uveitis (EMIU). J Autoimmun. 1998;11(2):111-8.
22. Yamaki K, Takiyama N, Itho N, Mizuki N, Seiya M, Sinsuke W, et al. Experimentally induced Vogt-Koyanagi-Harada disease in two Akita dogs. Exp Eye Res. 2005;80(2):273-80.
23. Otani S, Sakurai T, Yamamoto K, Fujita T, Matsuzaki Y, Goto Y, et al. Frequent immune response to a melanocyte-specific protein KU-MEL-1 in patients with Vogt-Koyanagi-Harada disease. Br J Ophthalmol. 2006;90(6):773-7.
24. Matsuzaki Y, Hashimoto S, Fujita T, Suzuki T, Sakurai T, Matsushima K, et al. Systematic identification of human melanoma antigens using serial analysis of gene expression (SAGE). J Immunother. 2005;28(1):10-9.
25. Yamada K, Senju S, Shinohara T, Nakatsura T, Murata Y, Ishihara M, et al. Humoral immune response directed against LEDGF in patients with VKH. Immunol Lett. 2001;78(3):161-8.
26. Yamada K, Senju S, Nakatsura T, Murata Y, Ishihara M, Nakamura S, et al. Identification of a novel autoantigen UACA in patients with panuveitis. Biochem Biophys Res Commun. 2001;280(4):1169-76.
27. Prasad PS, Levinson RD. In silico prediction of binding of putative antigenic peptides to HLA-DRB1 alleles in Vogt-Koyanagi-Harada disease. Clin Immunol. 2005;116(2):143-8.
28. Kondo I, Yamagata K, Yamaki K, Sakuragi S. [Analysis of the candidate antigen for Harada's disease[. J Jpn Ophthalmol Soc. 1994;98(6):596-603. Japanese.
29. Minoda H, Sakai J, Sugiura M, Imai S, Osato T, Usui M. [High inducibility of Epstein-Barr vírus replication in B lymphocytes in Vogt-Koyanagi-Harada disease]. Nippon Ganka Gakkai Zasshi. 1999;103(4):289-96. Japanese.
30. Sugita S, Takase H, Imai Y, Mochizuki M. Immunopathogenic mechanisms of a melanocyte-specific autoimmune disease: a cross-reaction between tyrosinase peptides and cytomegalovirus antigen. Invest Ophthalmol Vis Sci. 2002; 43:E-abstract 1529.
31. Chan CC, Palestine AG, Nussenblatt RB, Roberge FG, Benezra D. Anti-retinal auto-antibodies in Vogt-Koyanagi-Harada syndrome, Behcet's disease and sympathetic ophthalmia. Ophthalmology. 1985;92(8):1025-8.
32. de Smet MD, Yamamoto JH, Mochizuki M, Gery I, Singh VK, Shinohara T, et al. Cellular immune responses of patients with uveitis to retinal antigens and their fragments. Am J Ophthalmol 1990;110(2):135-42.
33. Yang P, Chen L, Zhou H, Zhong H, Wang H, Huang X, et al. Resistance of lymphocytes to Fas-mediated apoptosis in Behcet's disease and Vogt-Koyanagi-Harada syndrome. Ocul Immunol Inflamm. 2002:10(1):47-52.
34. Ohta K, Yoshimura N. Bcl-2 expression by CD4 T lymphocytes in Vogt-Koyanagi-Harada disease. Ocul Immunol Inflamm. 2002;10(2):93-103.
35. Chee SP, Luu CD, Cheng CL, Lim WK, Jap A. Visual function in Vogt-Koyanagi-Harada patients. Graefes Arch Clin Exp Ophthalmol. 2005;243(8): 785-90.
36. Bouchenaki N, Herbort CP. The contribution of indocyanine green angiography to the appraisal and management of Vogt-Koyanagi-Harada disease. Ophthalmology. 2001;108(1):54-64.
37. Bacsal K, Wen DS, Chee SP. Concomitant choroidal inflammation during anterior segment recurrence in Vogt-Koyanagi-Harada disease. Am J Ophthalmol. 2008;145(3):480-6.
38. Kawakami Y, Suzuki Y, Shofuda T, Kiniwa Y, Inozume T, Dan K, et al. T cell immune responses against melanoma and melanocytes in cancer and autoimmunity. Pigment Cell Res. 2000;13(Suppl 8):163-9.
39. Kobayashi H, Kokubo T, Takahashi M, Sato K, Miyokawa N, Kimura S, et al. Tyrosinase epitope recognized by an HLA-DR-restricted T-cell line from a Vogt-Koyanagi-Harada disease patient. Immunogenetics. 1998;47(5):398-403.
40. Yamaki K, Kondo I, Nakamura H, Miyano M, Konno S, Sakuragi S. Ocular and extraocular inflammation induced by immunization of tyrosinase related protein 1 and 2 in Lewis rats. Exp Eye Res. 2000;71(4):361-9.
 Address to correspondence:
Address to correspondence:
Francisco Max Damico
Rua Barata Ribeiro, 414 - Conj. 11
São Paulo (SP) CEP 01308-000
E-mail: [email protected]
Recebido para publicação em 10.11.2008
Aprovação em 05.01.2009
Uveitis Service, Hospital das Clínicas, Universidade de São Paulo Medical School. São Paulo (SP) - Brazil.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}
{kind=link}
{kind=link}
{kind=link}