Arq. Bras. Oftalmol. 2000; 63 (6): 10.1590/S0004-27492000000600005
Total: 3781
Fernando Antônio de Macedo Leal1; Ednaldo Atem Gonçalves2; Josilene de Carvalho Soares Liarth3; Fábio Martins Soares4
DOI: 10.1590/S0004-27492000000600005
RESUMO
Objetivo: A síndrome de Alport caracteriza-se por nefropatia hereditária, geralmente associada à surdez neurossensorial (Alport, 1927) e alterações oculares (Sohar, 1954). Estudaram-se as manifestações da síndrome em membros de uma mesma família, considerando aspectos clínicos e genéticos. Pacientes e Método: Foram submetidos a exame oftalmológico quinze indivíduos de uma família, dos quais quatro apresentavam síndrome de Alport. Os exames foram realizados na Clínica Oftalmológica do Hospital Getúlio Vargas - Universidade Federal do Piauí (HGV/UFPI). Resultados: Revelaram-se quatro indivíduos afetados pela síndrome de Alport, dos quais três apresentavam surdez moderada, lenticone anterior bilateral e nefropatia crônica, e um tinha manifestação renal isolada. Todos os pacientes eram brancos, do sexo masculino, com idade entre 12 e 25 anos. A acuidade visual dos olhos com lenticone variou de 20/50 a 20/100. Conclusões: A análise da família identificou três casos confirmados e um suspeito, sugerindo padrão de herança recessiva ligada ao X, não sendo possível afastar a herança autossômica dominante com penetrância incompleta.
Descritores: Síndrome de Alport; Lenticone
ABSTRACT
Purpose: Alport's Syndrome is characterized by hereditary nephritis, usually associated with neurosensitve deafness (Alport, 1927) and ocular disease (Sohar, 1954). Thus we intended to analize manifestations of the syndrome in a family, regarding clinical and genetical aspects. Patients and method: Fifteen patients of the same family of which four had Alport´s syndrome were submitted to ophthalmologic examination. The examinations were performed at the Ophthalmologic Clinic of HGV/UFPI. Results: Four patients with Alport´s syndrome were revealed, of which three had moderate deafness, anterior lenticonus and chronic nephropathy, and one of them had renal disease alone. All the patients were white, male aged 12 to 25 years. The visual acuity of eyes with lenticonus varied between 20/50 and 20/100. Conclusions: The family analysis identified three confirmed cases and a suspected one, sugesting a pattern of X-linked recessive inheritance, and it was not possible to exclude autosomal dominant inheritance with incomplete penetrance.
Keywords: Alport's syndrome; Lenticonus
INTRODUÇÃO
A Síndrome de Alport é caracterizada por nefropatia hereditária, frequentemente associada à surdez neurossensorial e alterações oculares. Em 1927, Alport descreveu a associação de distúrbios renais e auditivos, ao passo que Sohar, no ano de 1954, observou a presença de comprometimento ocular 1.
A nefrite é a alteração mais comum, iniciando-se geralmente na adolescência com proteinúria e/ou hematúria intermitentes. Apresenta curso crônico e progressivo para insuficiência renal, afetando principalmente indivíduos do sexo masculino 1, 2. A disacusia é tipicamente bilateral, simétrica e progressiva, sendo confirmada pelo exame audiométrico e instalando-se ainda na adolescência em 60% dos homens afetados e 40% das mulheres com a síndrome 1.
As alterações oculares ocorrem em 15% dos pacientes 2 e a presença de lenticone anterior, característica importante da doença, relaciona-se a um pior prognóstico sistêmico 1, 3. Outros achados incluem: arco senil, rotura da cápsula anterior do cristalino, catarata subcapsular, esferofacia, síndrome da dispersão pigmentar, heterocromia de íris, atrofia coroidiana, drusas do disco óptico, pigmentação macular e descolamento de retina 3.
A patogênese é ainda objeto de discussão. Em 1983, Govan sugeriu a existência de um defeito na biossíntese do colágeno na membrana basal do glomérulo e na cápsula lenticular 1. Parece ocorrer uma mutação gênica que levaria à produção alterada do colágeno do tipo IV, componente essencial da membrana basal de diversos órgãos, o que pode explicar a ocorrência de outros achados sistêmicos descritos, tais como: disfunção cerebral, polineuropatias, ictiose, e alterações de tireóide e paratireóides. Atualmente, defendem-se dois mecanismos básicos de herança: recessiva ligada ao cromossomo X e autossômica dominante 4.
Neste contexto, o presente trabalho propôs-se a estudar as manifestações da Síndrome de Alport em membros de uma mesma família, analisando os aspectos clínicos e genéticos envolvidos.
PACIENTES E MÉTODO
Foram submetidos a exame oftalmológico quinze indivíduos de uma família, dos quais quatro apresentavam manifestações clínicas sugestivas da Síndrome de Alport. Os exames foram realizados na Clínica Oftalmológica do Hospital Getúlio Vargas - Universidade Federal do Piauí (HGV/UFPI), no período de outubro de 1998 a março de 1999.
RESULTADOS
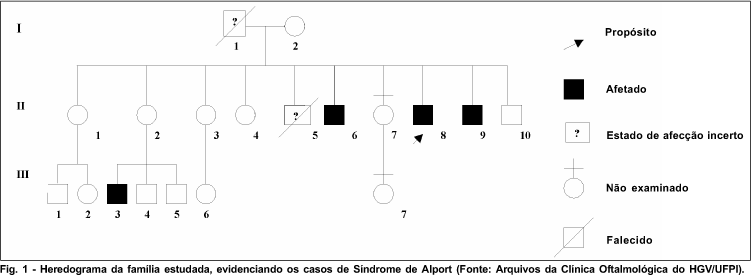
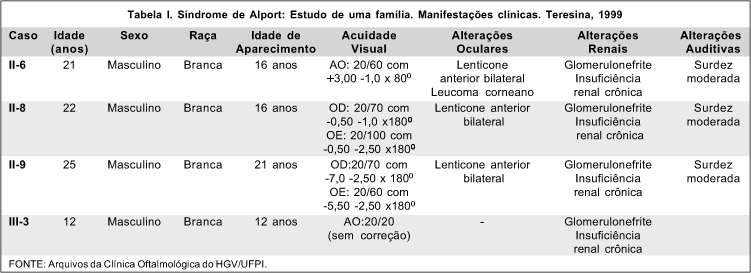
Após o exame dos quinze indivíduos da família, foi construído um heredograma (Figura 1). Foram identificados quatro indivíduos afetados pela Síndrome de Alport: II-6, II-8, II-9 e III-3. Os indivíduos II-6, II-8 e II-9 apresentavam lenticone anterior bilateral, surdez moderada e nefropatia crônica. O indivíduo III-3 apresentava o quadro renal isolado, isto é, sem alterações oculares ou auditivas associadas. Todos os pacientes eram brancos, do sexo masculino, com idade entre 12 e 25 anos. A acuidade visual dos olhos com lenticone variou de 20/50 a 20/100 (Tabela I).
DISCUSSÃO
Na família estudada, foram identificados três casos que apresentavam alterações renais, oculares e auditivas (II-6, II-8 e II-9), e um caso (III-3) que apresentava somente alteração renal. Durante a investigação, verificou-se história de óbitos em decorrência de doença renal do pai e um irmão dos pacientes da segunda geração, anteriores ao presente estudo (Figura 1). Não se acharam relatos na literatura em relação à preferência por grupos raciais, muito embora todos os pacientes do presente estudo sejam brancos.
O paciente II-8 era portador de insuficiência renal iniciada aos 16 anos, ao passo que os pacientes II-6, II-9 e III-3 apresentaram a mesma doença aos 16, 21 e 12 anos, respectivamente. Segundo Goldchmit e Almeida 1, as primeiras manifestações iniciam-se geralmente na adolescência, com quadro de hematúria e/ou proteinúria, havendo progressão para insuficiência renal ainda na adolescência, da segunda à quarta década ou até mesmo na quinta.
Os casos em questão apresentavam diagnóstico clínico de glomerulonefrite com conseqüente desenvolvimento de insuficiência renal crônica. A histologia renal caracteriza-se por alterações glomerulares e túbulo-intersticiais, sendo controversa a especificidade das mesmas. Com relação ao tratamento, o paciente II-8 submeteu-se a transplante renal em 1993, com sucesso, e os pacientes II-6 e II-9 encontravam-se em programa de hemodiálise. O paciente III-3 havia iniciado programa de diálise ambulatorial contínua em fevereiro/1999.
O exame audiométrico dos pacientes II-6, II-8 e II-9 revelou disacusia sensorial bilateral de grau moderado. A perda auditiva ocorre principalmente para os sons de alta frequência, é progressiva e geralmente desenvolve-se em concomitância à progressão da doença renal 2. Miller e colaboradores mostrou envolvimento do neuroepitélio vestibular e da cóclea, não havendo, porém, homogeneidade em relação aos achados histopatológicos 5.
Os pacientes II-6, II-8 e II-9 apresentavam lenticone anterior bilateral, ao passo que o paciente II-6 possuía ainda leucoma corneano. O lenticone, descrito inicialmente por Webster, em 1875 6, é uma anomalia da curvatura do cristalino, caracterizada por protrusão cônica quase sempre bilateral do pólo anterior e/ou posterior 7. À transiluminação, evidencia aspecto que lembra o de uma "gota de óleo na água", podendo ser pesquisado na retinoscopia, oftalmoscopia direta ou biomicroscopia com iluminação em campo vermelho (Figura 2). Tal fenômeno decorre de excessiva reflexão prismática na região axial, que impede a chegada ao olho do observador dos raios luminosos provenientes do fundo-de-olho do paciente 8. Existem relatos de pacientes com ataques de erosões epiteliais recorrentes e espontâneas 9, fato que poderia explicar a existência do leucoma do paciente II-6; todavia, as informações prestadas pelo mesmo não confirmaram a hipótese. Nenhum dos pacientes do estudo apresentava alteração vítreo-retiniana.
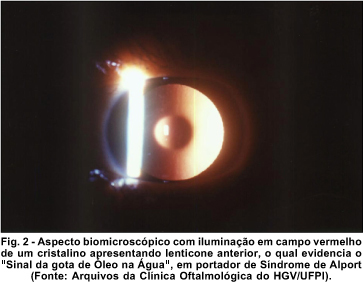
Em relação aos achados histopatológicos cristalinianos, há relatos de adelgaçamento da cápsula anterior na sua região central, com diminuição do número de células epiteliais na região do lenticone, além de alterações degenerativas nas fibras corticais. Existem muitas teorias que tentam explicar tais achados: retardo ou anomalia na separação da vesícula cristaliniana do ectoderma de superfície, fragilidade da cristalóide anterior, anomalia do sistema zonular, desequilíbrio entre a resistência da cápsula e a pressão intracristaliniana, trauma e perturbação metabólica 8.
As alterações oculares têm caráter progressivo, com redução da acuidade visual na maioria dos pacientes, devido às irregularidades do estado refrativo. Usualmente, o cristalino apresenta dupla refração: região central miópica e a periferia emétrope ou hipermetrópica, alterando a qualidade da visão. Com relação à terapêutica, "não existindo opacificação cristaliniana, o tratamento é óptico", ressaltaram Andrade Júnior e colaboradores em 1983 4. Contudo, com a modernização das técnicas de facectomia extracapsular e facoemulsificação, pacientes com lenticone sem opacidades cristalinianas podem ser visualmente reabilitados com a extração do cristalino transparente e implante de lente intra-ocular, como relatam Trindade e Katina 10. Segundo tais autores, a indicação da cirurgia de catarata não se baseia exclusivamente na acuidade visual e/ou presença de catarata, mas leva em consideração a alteração na qualidade da imagem e o conseqüente prejuízo para o exercício diário das atividades do paciente. Cresta relata um caso de um afetado que apresentou rotura espontânea da cápsula anterior e foi submetido à aspiração de massas e colocação de lente intra-ocular 11.
O exame desta família não permite a exata identificação do padrão de herança, pois não se tem certeza do estado de afecção de I-1. Mas, provavelmente o padrão de herança seja recessivo ligado ao cromossomo X, devido ao fato da mulher II-2 ser portadora de um gene recessivo no cromossomo X e ter passado este cromossomo X afetado para seu filho afetado III-3 (Figura 1). No entanto, não é possível afastar a herança autossômica dominante com penetrância incompleta 12.
O paciente III-3 possui somente alterações renais. Considerando que a síndrome inicia-se, em geral, na adolescência, com alterações renais e auditivas concomitantes 1 e as alterações oculares ocorrem em apenas 15% dos casos, não sendo essenciais ao diagnóstico da síndrome, admite-se que o paciente seja, de fato, portador da Síndrome de Alport com uma expressividade menor, pois não apresentou as alterações oculares ou auditivas.
CONCLUSÕES
O estudo das manifestações clínicas dos pacientes permitiu o diagnóstico de três casos na segunda geração e o diagnóstico de um caso provável na geração subseqüente, considerando a história familiar a evolução natural da doença. A análise do heredograma sugere padrão de herança recessiva ligada ao cromossomo X, mas não foi possível afastar a herança autossômica dominante com penetrância incompleta.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Goldchmit M, Almeida CV. Correlação entre o comprometimento renal e as alterações auditivas e oculares em pacientes portadores da Síndrome de Alport. Rev Bras Oftalmol 1990;49:45-9.
2. Nielsen CE. Lenticonus anterior and Alport's syndrome. Acta Ophthalmol 1978;56:518-30.
3. Singh DS, Bisht DB, Kapoor S, Sharma RN, Sankaran K, Majundar NK. Lenticonus in Alport's syndrome: A Family Study. Acta Ophthalmol 1977;55:164-9.
4. Andrade Júnior L, Andrade Neto L, Laércio W. Síndrome de Alport. Rev Bras Oftalmol 1983;42:61-8.
5. Miller GW, Joseph DJ, Cozard RL, McCabe BF. Alport's syndrome. Arch Otolaryngol 1970;92:419-32.
6. Paiva C. Lenticone Anterior (Lentiglobo). Rev Bras Oftalmol 1972;31:193-5.
7. Gonçalves FP, Baldrin T, Maldi V. Lenticone Anterior ¾ Lente de Contato. Rev Bras Oftalmol 1972;31:61-71.
8. Freitas VJ. Lenticone Anterior na Síndrome de Alport. Rev Bras Oftalmol 1976;35:171-7.
9. Rhys C, Snyers B, Pirson R. Recurrent Corneal Erosion Associated with Alport's Syndrome. Kidney Int 1997;52:208-11.
10. Trindade FC, Katina J. Lenticone Anterior na Sídrome de Alport: Reabilitação Visual após Facectomia Extracapsular em Cristalino Transparente com Implante de Lente Intra-ocular em Câmara Posterior. Rev Bras Oftalmol 1990;49:71-5.
11. Cresta BF, Silva ALB, Carvalho CA. Rotura Espontânea da Cápsula Anterior do Cristalino em Paciente com Síndrome de Alport. Arq Bras Oftalmol 1998;61:717-9.
12. Thompson & Thompson. Genética Médica. Rio de Janeiro: Guanabara Koogan, 1993.p.47-8.
Trabalho apresentado no XXX Congresso Brasileiro de Oftalmologia, no dia 06 de setembro de 1999, em Recife - PE.
(1) Residente do terceiro ano da Clínica Oftalmológica do Hospital Getúlio Vargas (HGV) / Universidade Federal do Piauí (UFPI).
(2) Professor-Adjunto de Oftalmologia do HGV/UFPI.
(3) Médica Oftalmologista da Clínica Oftalmológica do HGV/UFPI.
(4) Residente do primeiro ano da Clínica Oftalmológica do HGV/UFPI.
Endereço para correspondência: Dr. Fernando Antônio de Macedo Leal - Rua Coelho de Resende, 554 Sul - Teresina (PI) CEP 64.001-370.
Nota Editorial: Pela análise deste trabalho e por sua anuência sobre a divulgação desta nota, agradecemos a Dra. Juliana Maria Ferraz Sallum.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}
{kind=link}