Arq. Bras. Oftalmol. 2007; 70 (4): 10.1590/S0004-27492007000400026
Total: 2116
Marcela Cypel1; Rubens Belfort Jr.2; Nilva Moraes3; Cristina Muccioli4
DOI: 10.1590/S0004-27492007000400026
RESUMO
O linfoma não-Hodgkin primariamente intra-ocular é raro, e caracterizado por uma forma extranodal que pode envolver retina, espaço sub-retiniano, vítreo e nervo óptico. Ocorre independente ou associado ao linfoma do sistema nervoso central e freqüentemente na forma de uveíte de difícil tratamento. Esta forma de linfoma é um dos tumores intra-oculares mais desafiantes de se diagnosticar. Relatamos um caso de linfoma não-Hodgkin intra-ocular difuso de células grandes tipo B numa paciente de 47 anos e cuja manifestação inicial foi uma uveíte posterior; enfatizamos a importância de investigação detalhada e da avaliação sistêmica; pois esta é a forma mais freqüente de acometimento ocular. Com esse relato esperamos abordar os principais aspectos desde quadro clínico até diagnóstico chamando a atenção para essa doença que muitas vezes aparece inicialmente com sintomas vagos e inespecíficos.
Descritores: Uveíte; Neoplasias do sistema nervoso central; Linfoma, não Hodgkin; Síndromes paraneoplásicas; Relatos de casos
ABSTRACT
Ocular non-Hodgkin's lymphoma is a rare condition that can involve the retina, the vitreous and the optic nerve. It can occur alone or can be associated with lymphoma of the central nervous system and a frequent manifestation is a posterior uveitis of difficult treatment. This kind of ocular tumor is difficult and a challenge to diagnosis. We describe a case of non-Hodgkin's intraocular B-cell lymphoma in a 47-year-old woman who had a posterior uveitis as the first manifestation. We emphasize the importance of a careful investigation and of the general clinical examination since this is the most common type in the eye. We expect to call the attention to this disease that many times appears in an unspecific form with unspecific symptoms.
Keywords: Uveitis; Central nervous system neoplasms; Lymphoma, non-Hodgkin; Paraneoplastic syndromes
INTRODUÇÃO
Os linfomas não-Hodgkin (LNH) são um grupo heterogêneo de neoplasias do ponto de vista anatomopatológico e correspondem a 4% de todas as neoplasias humanas(1). Freqüentemente acometem o tecido linfóide (linfonodos e baço), mas podem apresentar manifestações extranodais em 25% dos casos, sendo os órgãos mais freqüentes o estômago, a pele, a cavidade oral, o intestino delgado e o sistema nervoso central (SNC)(1).
Linfoma intra-ocular primário (LIP) é raro(2-3) e caracterizado por uma forma extranodal que pode envolver retina, espaço sub-retiniano, vítreo e nervo óptico(3). É geralmente do tipo células B e do subtipo difuso de células grandes, sendo que alguns autores sugerem um imunofenotipo com origem em células centrais germinativas(2-3). O LIP ocorre independente ou associado ao linfoma do sistema nervoso central e freqüentemente na forma de uveíte de difícil tratamento. Esta forma de linfoma é um dos tumores intra-oculares mais desafiantes de se diagnosticar(3). O "padrão ouro" no diagnóstico dos LIP é feito a partir de biopsia vítrea com citologia e deve ser indicado em quadros de uveíte idiopática que sejam sugestivas de linfoma(3). O LIP do tipo células T é ainda mais raro e na maioria das vezes é secundário a um foco inicial cutâneo ou sistêmico de linfoma(2). O LIP de células B embora raro é a forma intra-ocular mais freqüente.
O objetivo deste artigo é relatar um caso de LIP difuso de células grandes tipo B, cuja manifestação inicial foi uma uveíte posterior e o diagnóstico somente realizado após manifestação neurológica.
RELATO DO CASO
Paciente do sexo feminino, 47 anos, espanhola, residente em São Paulo com história de baixa acuidade visual súbita no olho direito há 3 meses; negava dor, hiperemia, secreção ou outros sintomas. Sem tratamento prévio procurou um Centro de Referência em Oftalmologia.
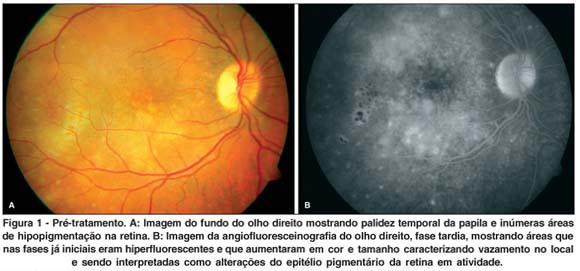
Ao exame oftalmológico apresentava acuidade visual de conta dedos a 1 metro olho direito (OD) e 20/20 olho esquerdo (OE). A única alteração ao exame de biomicroscopia era uma pequena quantidade de células vítreas no olho direito e o olho esquerdo apresentava-se sem alterações. Pressão intra-ocular dentro do normal em ambos os olhos. Exame fundoscópico do OD evidenciava uma palidez temporal da papila e inúmeras áreas de hipopigmentação difusas sugestivas de coroidite multifocal (Figura 1A). Ao exame de angiofluoresceinografia o OD mostrou áreas hiperfluorescentes nas fases já iniciais que aumentaram em cor e tamanho caracterizando vazamento no local e sendo interpretadas como alterações do epitélio pigmentário da retina (Figura 1B). O exame de campo visual mostrou uma baixa generalizada da sensibilidade no OD. O olho esquerdo era normal.
Foram solicitados exames de laboratório: HIV, PPD, FAN, sífilis, toxoplasmose; tomografia computadorizada de tórax, tomografia de crânio e avaliações sistêmicas com um clínico geral, um ginecologista e um neurologista.
A paciente retornou após 15 dias referindo piora do quadro. Todos os exames realizados estavam dentro dos padrões de normalidade, mas a paciente recusou-se a fazer a avaliação com o neurologista e a tomografia de crânio. Ao exame a acuidade visual do OD estava mantida em conta dedos a 1 metro e a do OE havia baixado para 20/30. À fundoscopia apresentava os mesmos pontos hipocrômicos dispersos na retina mas agora em maior quantidade e também presentes no OE. Neste momento optou-se por tratamento com corticóide via oral (Meticorten® 40 mg) na tentativa de melhorar acuidade visual da paciente. Foi feito nova solicitação de avaliação neurológica incluindo exame de líquor.
Após um mês a paciente evoluiu com visão inalterada do OD, OE 20/20 com melhora discreta do aspecto inflamatório da retina, iniciou-se a regressão da dose de Meticorten® até a sua suspensão; a paciente ainda se recusava a fazer os exames solicitados.
Após 4 meses do início dos sintomas a paciente evoluiu com quadro de convulsão e hemiparesia sendo então internada na neurologia onde foi realizada punção de líquor e tomografia computadorizada de crânio mostrando inúmeras lesões cerebrais próximas à região do tálamo sugestivas de linfoma. Foi submetida à cirurgia e biopsia de congelação que confirmou o diagnóstico de linfoma não-Hodgkin difuso de grandes células B infiltrando tecido nervoso central. O tratamento estipulado foi quimioterapia com vincristina, procantazina e metotrexato associado à radioterapia.
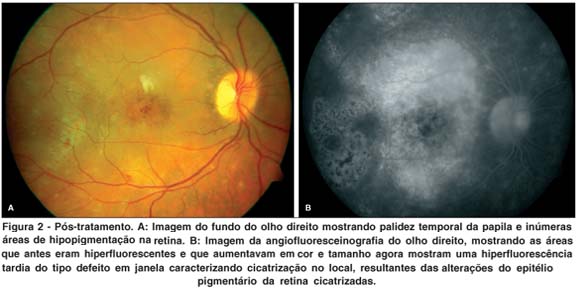
Após 1 ano de tratamento para o linfoma não-Hodgkin foi realizado avaliação oftalmológica que revelava visão de OD conta dedos a 1 metro e OE 20/20 e à fundoscopia apresentava cicatrização total das lesões, sem reação inflamatória em ambos os olhos. Tomografia computadorizada de crânio sem alterações.
DISCUSSÃO
O linfoma não-Hodgkin intra-ocular juntamente com a leucemia, síndrome de Richter's, melanoma uveal e retinoblastoma faz parte da classe das síndromes mascaradas as quais simulam uveíte inflamatória, mas são decorrentes de processos neoplásicos(4-7).
Dentre as causas de síndromes mascaradas é a causa mais comum descrita em 25% dos linfomas primários do SNC(4-6). O linfoma difuso de grandes células B é descrito como agressivo(1) e de prognóstico ruim principalmente quando linfoma primário do sistema nervoso central ou linfoma primário intraocular associado a acometimento do sistema nervoso central(3); alguns autores descrevem que independente do tratamento a sobrevida é de 12 a 18 meses(4-5); outros aumentam para 20 meses(7). No caso descrito neste artigo a paciente já se encontra com 19 meses do início dos sintomas e aparentemente sem sinais de recidiva tanto cerebral como ocular. Existe um outro relato de um caso também nacional no qual a confirmação diagnóstica foi de um linfoma uveal extranodal de células B sem envolvimento sistêmico foi realizada a enucleação do olho acometido e a paciente se encontra com 8 anos de evolução sem qualquer manifestação sistêmica ou necessidade de outros tratamentos(8).
Os sintomas oculares descritos na literatura são: baixa visual uni ou bilateral, floaters, uveíte anterior leve ou moderada, uveíte posterior leve (vitreíte) com borramento do nervo óptico. Nessa fase é difícil fazer o diagnóstico, pois se pensa normalmente em causa autoimune ou infecção; uma vez introduzido corticóide a inflamação diminui, mas não se mantém retornando e permanecendo resistente ao tratamento. Quando acometimento da retina o diagnóstico é mais fácil, mas a ausência de sinais de acometimento do SNC pode atrapalhar o diagnóstico. O sinal mais característico são lesões amarelo-esbranquiçadas e pouco elevadas sub-retinianas, pode ter descolamento do epitelio pigmentário da retina (EPR) e células vítreas; precipitados ceráticos e sinéquias podem não aparecer. Os achados são normalmente bilaterais e assimétricos(2). Sintomas do sistema nervoso central: diplopia, paresias, ataxias alterações de comportamento e aumento da pressão intracraniana(4-7). Uma vez identificado o linfoma primário do SNC é mandatório uma investigação detalhada do SNC pois a taxa de mortalidade nestes casos é maior(8). O caso acima descrito possui quase todos os sintomas descritos na literatura.
O diagnóstico padrão é feito pela punção de fluido ou tecido intra-ocular(2,4-7) (vítreo, coróide). Outras formas de diagnóstico são: punção do líquor (meio diagnóstico usado no caso aqui descrito), tomografia computadorizada, ressonância magnética, biopsia incisional intra-orbitária e até cerebral(6). Importante lembrar que estudos patológicos descrevem que as células neoplásicas têm uma distribuição variável no vítreo, podendo dar um falso negativo se a punção for feita num local de baixa concentração. Outras causas de falso negativo pela biopsia podem ser: conservação inadequada do material colhido, a corticoterapia que pode diminuir a concentração de células inflamatórias e às vezes as células neoplásicas se encontram sub-retinianas ficando no vítreo apenas as inflamatórias.
Se após a primeira consulta oftalmológica a paciente tivesse seguido as orientações dadas e realizado tanto a tomografia de crânio como a avaliação com o neurologista provavelmente o diagnóstico teria sido feito um mês e meio antes o que segundo descrito por vários autores(3,6) interfere diretamente no prognóstico.
Quanto ao diagnóstico diferencial devem ser consideradas as uveítes verdadeiras e esclerites: coroidite multifocal infecciosa, necrose de retina, toxoplasmose ocular, histoplasmose ocular, citomegalovírus, HIV(7).
O tratamento preconizado é a quimioterapia sistêmica, a radioterapia é usada nos casos de acometimento ocular, uma das drogas usadas com bons resultados é o metotrexate(3-5,7), o caso aqui descrito também segue essas recomendações.
Embora o LIP difuso de células grandes tipo B seja uma forma rara, nós oftalmologistas devemos tê-la sempre em mente, pois em se tratando de linfoma é a forma mais freqüente de acometimento ocular. Com esse relato de caso ressaltamos a necessidade da investigação diagnóstica, especialmente em casos oculares que apresentam associação com doenças sistêmicas. Salientamos a importância da confirmação do diagnóstico e a pronta instituição do tratamento que contribui para melhor prognóstico visual e sistêmico e maior chance de sobrevida.
REFERÊNCIAS
1. Colleoni GW, Oliveira JS. Linfomas. In: Prado FC, Ramos JA, Valle JR. Atualização Terapêutica. 22a ed. São Paulo: Artes Médicas; 2005. p.831-46.
2. Coupland SE, Anastassiou G, Bornfeld N, Hummel M, Stein H. Primary intraocular lynfoma of T-cell type: report of a case and review of the literature. Graefes Arch Clin Exp Ophtalmol. 2005;243(3):189-97.
3. Coupland SE, Heimann H. Primary intraocular lymphoma. Ophthalmologe. 2004;101(1):87-98.
4. Dugel PU, Thach AB. Ocular neoplasms related to human immunodeficiency virus. In: Yanoff M, Duker JS, editors. Ophthalmology. 2nd ed. St. Louis: Mosby; 2004. p.1229-32.
5. Read RW. Masquerade syndromes: neoplasms. In: Yanoff M, Duker JS, editors. Ophthalmology. 2nd ed. St. Louis: Mosby; 2004. p.1233-38.
6. Kaushik S, Gupta V, Singh R, Vasishta RK, Rajwanshi A, Grupta A. Disseminated metastasis following periampullary câncer resection. Indian J Ophthalmol. 2005;53(1):57-9.
7. Read RW, Zamir E, Rao NA. Neoplastic masquerade syndromes. Surv Ophthalmol. 2002;47(2):81-124.
8. Coutinho AB, Muccioli C, Martins MC, Belfort Jr R, Sant'Anna AE, Burnier Jr MN. Extranodal B-cell lymphoma of the uvea: a case report. Can J Ophthalmol. 2005;40(5):623-6.
Endereço para correspondência:
Marcela Cypel
Rua Simão Álvares, 1015
São Paulo (SP) - CEP 05417-030
E-mail: [email protected]
Recebido para publicação em 22.05.2006
Última versão recebida em 29.10.2006
Aprovação em 07.11.2006
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Zélia Maria da Silva Corrêa sobre a divulgação de seu nome como revisor, agradecemos sua participação neste processo.
Trabalho Realizado no Instituto da Visão Departamento de Oftalmologia da Universidade Federal São Paulo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.