Arq. Bras. Oftalmol. 2006; 69 (6): 10.1590/S0004-27492006000600027
Total: 2812
Ana Karina Santiago de Medeiros Lima1; Carlos Alexandre de Amorim Garcia2; Marco Antônio Rey de Faria3; Uchoandro Bezerra Costa Uchoa4
DOI: 10.1590/S0004-27492006000600027
RESUMO
A síndrome de Cogan é entidade multissistêmica rara caracterizada por ceratite intersticial associada à disfunção áudio-vestibular e possível surdez irreversível classificada em duas formas clínicas: típica e atípica. Há discordância na literatura quanto à presença de acometimento corneano na forma atípica. Uma paciente de 32 anos queixando-se de hiperemia e dor ocular, fotofobia e baixa da acuidade visual no olho direito, associada à perda súbita de audição à esquerda, vômitos, diarréia, oligúria, dor na orofaringe e febre. História prévia de semelhante acometimento do olho esquerdo e audição direita. Havia intensa hiperemia conjuntival, esclerite nodular, episclerite e infiltrados circulares no estroma corneano. A paciente recebeu pulsoterapia com metilprednisolona e ciclofosfamida. Evoluiu com grande melhora ocular, porém com resposta auditiva pobre. O caso reportado pode constituir forma típica da síndrome de Cogan (de acordo com autores que defendem o não-acometimento corneano na forma atípica) com alguns achados característicos da forma atípica ou um caso da forma atípica da síndrome de Cogan (para aqueles que defendem o acometimento corneano na forma atípica). O diagnóstico diferencial também é discutido.
Descritores: Doenças da córnea; Esclerite; Perda auditiva; Doença de Meniere; Artrite; Síndrome; Doenças vestibulares; Surdez; Imunossupressores; Relatos de casos [tipo de publicação]
ABSTRACT
Cogan's syndrome is an unusual multisystemic disease characterized by intersticial keratitis in association with vestibuloauditory dysfunction and possible irreversible deafness, classified into 2 clinical types: typical and atypical. There is disagreement in the literature about corneal disease in the atypical variety. A 32-year-old woman complaining of ocular hyperemia and ocular pain, photophobia and visual acuity loss in the right eye associated with sudden left hearing loss, vomiting, diarrhea, oliguria, oropharynx pain and fever. Previous history of similar disease in left eye and right hearing. There was intense conjunctival hyperemia, nodular scleritis, episcleritis, and circular infiltrates in the corneal stroma. The patient received pulse-therapy with methylprednisolone and cyclophosphamide. She exhibited significant ocular improvement but poor hearing results. The reported case may be a typical Cogan's syndrome (according to authors that assert the non-existence of corneal disease in the atypical type) with some findings characteristic of the atypical type or an atypical Cogan's syndrome (for those asserting that it is a corneal disease). Differential diagnosis is also discussed.
Keywords: Corneal diseases; Scleritis; Hearing loss; Meniere's disease; Arthritis; Vestibular diseases; Deafness; Immunosuppressive agents; Case reports [publication type]
INTRODUÇÃO
Síndrome de Cogan é uma entidade clínica rara, caracterizada por ceratite intersticial não-sifilítica associada a disfunção áudio-vestibular (perda de audição, vertigem e "tinnitus") súbita(1) e bilateral na maioria dos casos(2). Geralmente não resulta em perda visual permanente, porém este aspecto é importante para o diagnóstico precoce e prevenção de surdez irreversível(1).
Inicialmente descrita como entidade clínica em 1945 por David Cogan, ocorre em adultos jovens (20-40 anos) e ainda não há dados de incidência ou prevalência pelo número limitado de pacientes documentados(1,3-5). A etiologia e fisiopatologia desta entidade permanecem desconhecidas(6), contudo há associação com infecção de vias aéreas superiores precedendo o quadro(7) e hipóteses de prováveis agentes virais(1). Outros acreditam que se trata de uma condição sistêmica auto-imune(4) associada a achados como aumento de velocidade de hemossedimentação (VHS), leucocitose com neutrofilia, variações de níveis séricos de complemento e imunoglobulinas, aumento dos níveis de linfócitos T e B, além de evidências de envolvimento inflamatório em pele, músculos, fígado(1) e grandes vasos (aorta)(1,4).
Há duas formas clínicas: a forma típica, ocorre em 90% dos casos, originalmente descrita com achados corneanos clássicos (infiltrados corneanos granulares no estroma profundo(1) e predominantemente próximos ao limbo, geralmente resultando em neovascularização estromal), associados à disfunção áudio-vestibular(1,7). A forma atípica cursa com inflamação ocular ou orbitária, associado à disfunção áudio-vestibular(1) e maior incidência de sintomas sistêmicos, principalmente relacionados à vasculite(7). Há discordância na literatura quanto à presença de acometimento corneano na forma atípica da síndrome(1,7-8).
RELATO DE CASO
Paciente de 32 anos, sexo feminino, brasileira, leucoderma, procedente de Natal - RN, foi atendida no serviço de oftalmologia do Hospital Universitário Onofre Lopes no dia 17/08/2004, com queixas de hiperemia, dor ocular, fotofobia e baixa da acuidade visual (AV) no olho direito (OD) que haviam iniciado 15 dias antes. Relatava perda súbita de audição à esquerda há 10 dias, vômitos freqüentes, oligúria, dor na orofaringe, diarréia há 3 dias e febre nos 2 dias iniciais do quadro. Em uso tópico ocular de ofloxacino e cetorolaco de trometamina, tendo ainda, feito uso tópico de gatifloxacino e cefalexina via oral (VO), sem apresentar melhora no quadro clínico. Afirmava semelhante acometimento de olho esquerdo (OE) e audição direita há 1ano e 3 meses. Hipertensa sem uso de drogas anti-hipertensivas. Negava outras comorbidades ou uso de medicamentos.
Ao exame, apresentava AV sem correção (S/C) 20/125 (+1) em OD e 20/60 em OE. À biomicroscopia, apresentava no OD intensa hiperemia conjuntival, esclerite nodular inferior, episclerite, infiltrados circulares no estroma corneano, flare e células (1+/4+) na câmara anterior. No OE havia hiperemia conjuntival moderada, infiltrado no estroma corneano e raras células na câmara anterior. Ainda, apresentava ausência de áreas corantes a fluoresceína e fundoscopia sem anormalidades em ambos os olhos (AO).
Prescrito para AO prednisolona colírio 2/2 hs e prednisona VO 60 mg/dia. Solicitados exames e encaminhada à clínica médica e otorrinolaringologia para elucidação do quadro sistêmico.
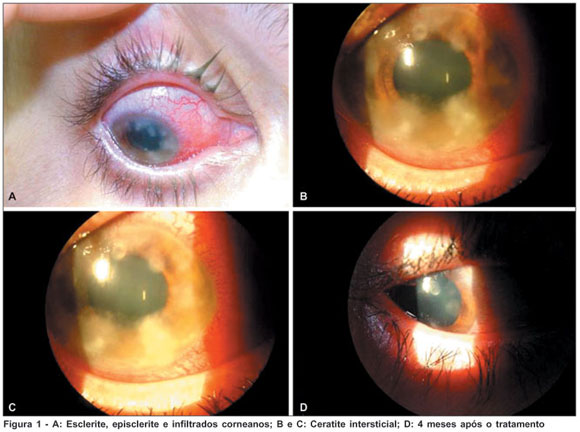
Dois dias após, já havia melhora acentuada do quadro ocular e aumento da dor em orofaringe.
Exames: VDRL não reativo; teste do látex negativo; leucograma 10.500/mm3 (neutrófilos= 67,3%, linfócitos= 20,8%, monócitos= 8,9%, eosinófilos= 1,7%, basófilos= 1,4%); plaquetas 500.000/mm3; dosagens aumentadas de transaminase glutâmica oxalacética (TGO) 49 U/L e transaminase glutâmica pirúvica (TGP) 89 U/L; tempo de tromboplastina parcial ativada (TTPA) normal.
Tomografia crânio-encefálica (TC) normal, audiometria revelava disacusia neurossensorial profunda bilateral com resposta até 4,0 KHz (90-100 dB) para ouvido esquerdo e sem resposta a estímulos sonoros 110 dB para ouvido direito.
Evoluiu com contínua melhora do quadro ocular e sistêmico com lesões corneanas em regressão (persistência apenas perifericamente) e após dois meses as lesões corneanas estavam cicatrizadas (nubéculas) em AO. Relatava discreta melhora auditiva. Realizou por duas vezes pulsoterapia em intervalo mensal, com metilprednisolona 1 g/3 dias e ciclofosfamida 1 g; estava em uso de prednisona 80 mg/dia e desmame de prednisolona colírio. Houve melhora do leucograma (14,6 K/uL) e normalização das dosagens de TGO e TGP, tendo sido iniciado desmame lento da prednisona VO.
Em 23/02/2004 retorna sem queixas oculares, referindo não ter melhorado audição, mesmo com uso de prótese auditiva. Olho calmo, sem reação de câmara anterior e leves opacidades corneanas estromais (nubéculas) mais evidentes na periferia do OD. PIO 15 mmHg no OD e 16 mmHg no OE. AV C/C 20/25 (+1) no OD e 20/30 (+2) no OE. Prednisolona colírio foi suspenso em 12/2004. Encontrava-se em uso de prednisona 10 mg/dia VO, cujo desmame foi mantido. A paciente foi orientada a retornar para acompanhamento após 3 meses.
DISCUSSÃO
A síndrome de Cogan é uma doença multissistêmica que em sua forma típica se caracteriza por ceratite intersticial não-luética e irite, associadas a sintomas semelhantes à doença de Ménière (náuseas, vômitos, vertigens e perda progressiva da audição)(7). Ainda não se evidenciou se seus sinais e sintomas representam uma entidade clínica distinta ou se são manifestações de outras desordens sistêmicas(9).
O caso reportado constitui uma forma típica da síndrome de Cogan para autores que defendem o não-acometimento corneano na forma atípica(1), com alguns achados da forma atípica como disfunção hepática (evidenciada por alterações de enzimas hepáticas), diarréia, episclerite, esclerite nodular inferior. Para os que defendem o eventual acometimento corneano(7-8), e conforme os critérios de Haynes constitui um caso da forma atípica(10). O achado de nodulação escleral impõe diagnóstico diferencial com poliarterite nodosa (esclerite necrosante), descartado com base na clínica áudio-vestibular da paciente(7). Outros possíveis diagnósticos diferenciais também foram excluídos, como sífilis (VDRL negativo), doenças reumáticas como artrite reumatóide (teste do látex negativo) e doença neurossensorial central (TC sem anormalidades), ototoxicidade medicamentosa (sem uso de salicilatos, diuréticos ou metais pesados)(1), granulomatose de Wegener(7) (sem sinais de vasculite sistêmica ou comprometimento intenso de trato respiratório superior com dor e secreção nasal purulenta ou sanguinolenta), síndrome de Vogt-Koyanagi-Harada (ausência de alterações fundoscópicas), doença de Ménière (sem acometimento ocular relatado)(1). Processos infecciosos também foram descartados: rubéola (sem rash cutâneo típico ou adenopatia cervical), parotidite (envolvimento de parótida e perda auditiva associada à meningite)(1), infecção por varicela-zoster(11) (sem acometimento dermatológico). Há outros diagnósticos diferenciais que devem ser aventados, como doença de Lyme, tuberculose e infecção por Clamydia(1).
Achados laboratoriais reforçaram a hipótese de síndrome de Cogan: como leucocitose com neutrofilia relativa e aumento de enzimas hepáticas, os quais foram normalizados com terapia adequada. A audiometria confirmou o severo défict neurossensorial, sem melhora com tratamento, típico da doença. História de infecção prévia de vias aéreas superiores é descrita no caso, como em 42% dos pacientes com forma típica da doença e em 65% das formas atípicas(1). Outros achados sistêmicos compatíveis são febre, cefaléia, dor cervical e na orofaringe (50-75%), diarréia (30%)(1). Os típicos achados oculares como lacrimejamento, fotofobia, hiperemia, baixa de acuidade visual, infiltrados corneanos em estroma profundo, pequenos (aproximadamente 1,0 mm de diâmetro)(1) e periféricos(1,7) estão de acordo com achados descritos na literatura. A pronta resposta do quadro ocular a corticoterapia tópica, a melhora dos sintomas sistêmicos a corticoterapia sistêmica e uso de agentes citotóxicos(5), com pobres respostas e prognóstico auditivo(5,12) é um típico resultado dos casos descritos, tendo sido documentado surdez bilateral em 40% destes(13). Tal fato reforça a importância do diagnóstico precoce de entidade com acometimento ocular associado a achados da doença de Mèniére como tentativa de minimizar seqüelas através da pronta administração de terapia imunossupressora(14).
REFERÊNCIAS
1. Kowal VO, Laibson PR. Keratitis due to Cogan's syndrome. Ophthalmol Clin North Am. 1994;7:649-56
2. Zierhut M, Schlote T, Stubiger N, Daikeler T, Kotter I, Bless D, Koitschev A. [Cogan I syndrome: clinical aspects, therapy and prognosis] Ophthalmologe. 2000;97(3):197-202. German.
3. Cogan DG. Syndrome of nonsyphilitic interstitial keratitis and vestibuloauditory symptoms. Arch Ophthalmol. 1945;33:144-9.
4. Oldenski R. Cogan syndrome: autoimmune-mediated audiovestibular symptoms and ocular inflammation. J Am Board Fam Pract. 1993;6(6):577-81.
5. Merle H, Trode M, Smadja D, Numeric P, Richer R, Jallot-Sainte-Rose N. [Interstitial keratitis and Cogan syndrome] J Fr Ophtalmol. 1995;18(1):50-4. French.
6. Pino Rivero V, Marcos Garcia M, Trinidad Ruiz G, Gonzalez Palomino A, Pardo Romero G, Trinidad Ramos G, et al. [Cogan sindrome. Audio-vestibular findings in a patient and literature review]. An Otorrinolaringol Ibero Am. 2004;31(6):601-7. Spanish.
7. Belfort Jr R, Kara-José N. Córnea: clínica-cirúrgica. São Paulo: Roca. 1996.
8. Pleyer U, Baykal HE, Rohrbach JM, Bohndorf M, Rieck P, Reimann J, Kuhnt P, et al. [Cogan I syndrome: too often detected too late? A contribution to early diagnosis of Cogan I syndrome]. Klin Monatsbl Augenheilkd. 1995;207(1):3-10. German.
9. Laude A, Fauchais AL, Hachulla E, Viget N, Lambert M, Michon-Pasturel U, et al. [Cogan syndrome or sarcoidosis?]. Rev Med Interne. 2002;23(8):720-3. French.
10. Haynes BF, Kaiser-Kupfer MI, Mason P, Fauci AS. Cogan syndrome: studies in thirteen patients, long-term follow-up, and a review of the literature. Medicine (Baltimore). 1980;59(6):426-41.
11. Schedler MG, Bartylla M. [Retrospective and prospective studies of patients with Cogan I syndrome]. Laryngorhinootologie. 1994;73(12):662-6. German.
12. McDonald TJ, Vollertsen RS, Younge BR. Cogan's syndrome: audiovestibular involvement and prognosis in 18 patients. Laryngoscope. 1985;95(6):650-4.
13. Wilder-Smith E, Roelcke U. [Cogan syndrome. Case report, review of the literature, therapy]. Laryngorhinootologie. 1991;70(2):90-2. German.
14. Orsoni JG, Zavota L, Pellistri I, Piazza F, Cimino L. Cogan syndrome. Cornea. 2002;21(4):356-9.
Endereço para correspondência:
Carlos Alexandre de Amorim Garcia
Rua Ceará Mirim, 316
Natal (RN) CEP 59020-240
E-mail: [email protected]
Recebido para publicação em 16.11.2005
Última versão recebida em 20.03.2006
Aprovação em 24.05.2006
Trabalho realizado na Disciplina de Oftalmologia da Universidade Federal do Rio Grande do Norte - UFRN - Natal (RN) - Brasil

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.