Arq. Bras. Oftalmol. 2006; 69 (6): 10.1590/S0004-27492006000600018
Total: 2518
Vanderson Glerian Dias1; Michel Castilho Salem2; Caio César Benetti Filho3
DOI: 10.1590/S0004-27492006000600018
RESUMO
OBJETIVOS: Diagnosticar, avaliar e descrever os achados clínico-genéticos e oftalmológicos de pacientes com síndrome de Stickler tipo II de uma mesma família. MÉTODOS: Todos os pacientes com alterações oftalmológicas foram submetidos à radiografia de mãos e punhos para idade óssea e posteriormente analisados pelo exame clínico-genético. O diagnóstico de síndrome de Stickler foi dado mediante análise clínica e correlação com o perfil metacarpofalangeano visualizado na radiografia. RESULTADOS: Síndrome de Stickler tipo II foi comprovada em 11 pacientes. Os achados oculares mais importantes foram: alta miopia (80%), subluxação do cristalino (70%), exotropia (50%) e anomalias vítreo-retinianas (80%) incluindo vazio vítreo (50%). O exame clínico-genético revelou que 30% dos pacientes apresentavam micrognatia, 50% hipoacusia, 40% depressão nasal e 60% palato alto. Hipermotilidade articular e dedos longos foram demonstrados em 7 casos (70%) e artropatia esteve presente em 3 pacientes (30% dos casos). CONCLUSÕES: O diagnóstico da síndrome de Stickler é difícil devido à variabilidade fenotípica e a existência de outras síndromes genéticas com características semelhantes. As radiografias de mão e punho são de particular importância no diagnóstico desta síndrome.
Descritores: Doenças do tecido conjuntivo; Anormalidades craniofaciais; Oftalmopatias hereditárias; steocondrodisplasias; Síndrome; Avaliação; Linhagem
ABSTRACT
PURPOSE: To diagnose, evaluate and describe the clinical, genetic and ophthalmic characteristics of a family with type II Stickler syndrome. METHODS: X-rays for bone age, clinical and genetic evaluation were performed in all patients with ocular alterations. The Stickler syndrome diagnosis was established after correlating these examinations. RESULTS: Type II Stickler syndrome was found in 11 patients. The most important ocular findings were: high myopia (80%), lens subluxation (70%), exotropia (50%) and vitreoretinal abnormalities (80%) including vitreous cavity (50%). The clinical genetic examination disclosed that 30% of the patients had micrognathia, 50% hearing loss, 40% nasal depression and 60% high palate. Seven cases had articular hypermotility and long fingers and arthropathy was present in 3 cases. CONCLUSION: Diagnosis of the Stickler syndrome is difficult due to its phenotypic variability and the existence of other genetic syndromes with similar characteristics. Hand and wrist radiographs are of particular importance in the diagnosis of this syndrome.
Keywords: Connective tissue diseases; Craniofacial abnormalities; Eye diseases; Osteochondrodysplasias; Syndrome; Evaluation; Pedigree
INTRODUÇÃO
Em 1965, Stickler et al., descreveram uma nova entidade dominante constituída de miopia iniciando nas primeiras décadas de vida e resultando em descolamento de retina e cegueira(1).
Atualmente a síndrome de Stickler (SS) é classificada em três tipos, de acordo com o gene no qual ocorre a mutação: tipo I, por mutação no gene COL2A1 (OMIM#108300)(2-5); tipo II, por mutação no gene COL11A1 (OMIM#604841)(6-8); e tipo III, por mutação no gene COL11A2 (OMIM# 184840)(9-10).
Várias são as alterações clínicas da SS, dentre as quais: problemas cardíacos(11), faciais(12-13), perda auditiva(12,14), além de alterações orais(15) e ósseas(13-14). Em relação às alterações oculares, a SS tipo I caracteriza-se por apresentar descolamento de retina, cegueira(1), glaucoma(16), catarata, alterações vítreas(17) e alta miopia(1,17-18). A SS tipo II também pode apresentar descolamento de retina, catarata e glaucoma, além de miopia antes de 6 anos de idade, sinerese vítrea(16), degeneração vítrea, atrofia difusa do epitélio pigmentar da retina(15,17), dentre outras. Já a SS tipo III não está relacionada a achados oculares.
A síndrome de Stickler tipo II, também conhecida como SS vítreo tipo 2, tem como marca principal uma anormalidade da arquitetura vítrea, sendo esta alteração, um dos pré-requisitos para o diagnóstico, além de miopia congênita de alto grau não progressiva(7). Em se tratando de achados sistêmicos, as pessoas com SS tipo II caracterizam-se por apresentarem micrognatia, perda auditiva neuro-sensorial, alterações orais, nasais e faciais, dedos longos, hipermobilidade das articulações, artropatia após a terceira década de vida(19).
Devido às várias mutações descritas na literatura, novos genes provavelmente relacionados à SS e a dificuldade clínica no diagnóstico diferencial, muitos profissionais médicos não fazem o diagnóstico correto dessa síndrome(7-8,20-24).
Nosso objetivo é diagnosticar, avaliar e descrever os achados clínico-genéticos e oftalmológicos de pacientes com síndrome de Stickler tipo II de uma mesma família, com o intuito de ajudar os oftalmologistas na distinção sindrômica.
MÉTODOS
Foram realizados exames oftalmológicos em 25 pessoas de uma família composta por 33 indivíduos (incluindo 7 óbitos e um paciente que não compareceu ao atendimento).
O exame oftalmológico baseou-se em: acuidade visual (AV) com a melhor correção pela tabela de Snellen; ectoscopia; exame de motilidade ocular; tonometria, feita com tonômetro de aplanação marca Goldmann; biomicroscopia do segmento anterior, realizada com lâmpada de fenda marca Shin-Nipon modelo SL-101; e mapeamento de retina, realizado com oftalmoscópio indireto marca Neitz modelo IOH.
Dos 25 pacientes examinados oftalmologicamente, 12 foram encaminhados para realizarem radiografia de mãos e punhos e avaliação clínica com geneticista conforme critérios estabelecidos abaixo para diagnóstico da síndrome de Stickler tipo II (Figura 1).
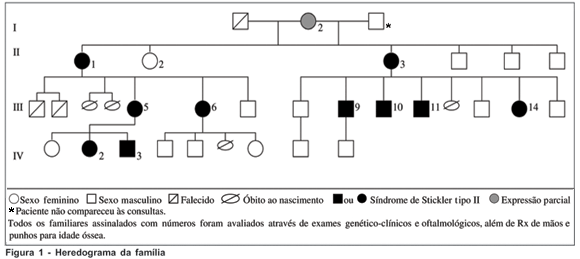
Como critério de encaminhamento para realizar radiografia de mãos e punhos para idade óssea e posterior exame clínico-genético, foi considerado pelo menos uma das seguintes alterações oculares: miopia de início antes de 6 anos de idade, atrofia difusa do epitélio pigmentar da retina, anomalias congênitas do vítreo, descolamento regmatogênico da retina, degeneração tipo "Lattice", glaucoma ou catarata(15-17,19).
Em relação ao exame clínico-genético, considerou-se caso de SS tipo II todo paciente que apresentava pelo menos três das seguintes alterações fenotípicas: micrognatia, perda auditiva neuro-sensorial, alterações orais, nasais e faciais, dedos longos, hipermobilidade das articulações, artropatia após a terceira década de vida(12-15,19).
O principal critério adotado para a diferenciação da síndrome de Stickler em relação à síndrome de Marfan, além dos dados oftalmológicos e clínico-genéticos, foi a análise das radiografias de mãos e punhos através do método descrito abaixo:
Padrão do perfil metacarpofalangeano (PPMF): feita através da técnica dos escores Z, na qual todos os ossos longos da mão são medidos em seu comprimento máximo (exceção feita ao pequeno processo na epífise proximal do 3º metacarpiano). As epífises dos ossos, quando presentes, são incluídas. Determina-se a seguir quanto cada um desses ossos se distancia dos valores médios de referência para idade e sexo do paciente. Essa dispersão em torno da média é dada em unidades de desvio-padrão ou escores Z segundo a fórmula(25):
Zi,s,o = [ (X0 - M i,s,o) / S i,s,o]
Onde:
X = medida do osso em milímetros (mm).
M = média das medidas normais para o mesmo osso, na mesma idade e sexo.
O = desvio-padrão das medidas normais para o mesmo osso, na mesma idade e sexo.
i,s,o = variável expressa para idade (i), sexo (s) e osso (o).
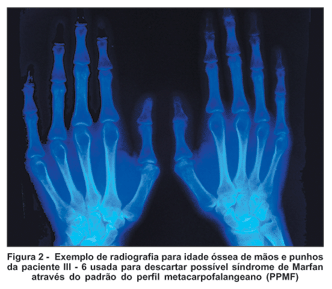
Essa técnica torna possível visualizar alterações individuais, em segmento, raios, ou ainda na proporção entre os ossos do esqueleto da mão. Em algumas síndromes os gráficos de PPMF assumem confrontações bem típicas, embora não patognomônicas, e podem ajudar bastante no diagnóstico(25).
O diagnóstico de síndrome de Stickler tipo II foi constatado mediante confrontação dos achados oftalmológicos, clínico-genéticos e dos exames radiológicos mediante análise do PPMF conforme critérios indicados acima.
Após a confirmação diagnóstica os pacientes foram analisados e as alterações clínico-genéticas e oftalmológicas foram descritas e comentadas.
RESULTADOS
Em relação aos dados demográficos, 6 pacientes com SS tipo II (60% dos casos) eram do sexo feminino e 4 (40% dos casos), do sexo masculino. A raça parda esteve presente em 80% dos casos (8 pacientes) e a raça branca, em apenas 20% (2 pacientes).
Comprovou-se SS tipo II em 10 pacientes (40%), uma paciente sem alterações condizentes com SS tipo II (4%) e a matriarca (4%) apresentando expressão parcial, sem comprometimento vítreo e com poucas alterações fenotípicas (Quadro 1).
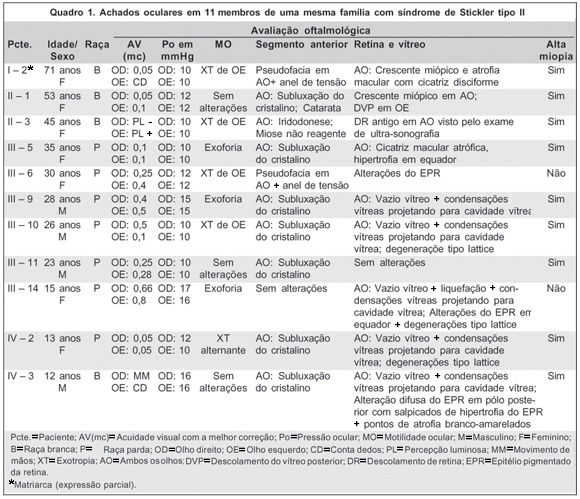
Miopia foi constatada em 8 pacientes (80% dos casos), tendo média refrativa de -11,75 dioptrias esféricas (DE), alterações vítreo-retinianas foram encontradas em 8 pacientes (80% dos casos), baixa da AV, em todos os pacientes com SS tipo II e também na matriarca, a qual apresentava crescente miópico e atrofia macular disciforme em AO, além de pseudofacia em AO. Das alterações vítreas, a mais comumente encontrada foi o vazio vítreo com projeção de condensação vítrea (5 pacientes - 50% dos casos), seguida pelas alterações da retina tipo lattice em 3 pacientes (30% dos casos) e alteração do epitélio pigmentar da retina (EPR) também em 3 casos (30%). Crescente miópico, descolamento do vítreo posterior (DVP), liquefação vítrea, descolamento de retina antigo (visto por meio da ultra-sonografia devido à dificuldade de dilatação pupilar) e cicatriz macular atrófica foram identificados em 10% dos pacientes com SS tipo II. Somente 1 paciente (10%) não apresentou alteração vítreo-retiniana (Quadro 1).
Dentre as alterações do segmento anterior a mais importante foi a subluxação do cristalino aparecendo em 70% dos casos (7 pacientes). Iridodonese, miose não-reagente e catarata nuclear foram constatadas em uma única paciente (10%). Um caso com SS tipo II foi submetido à cirurgia de facoemulsificação com implante de anel de tensão subcapsular, além da matriarca. Somente um paciente não apresentou alterações no segmento anterior.
O exame de motilidade ocular constatou exotropia (XT) em 4 pacientes com a síndrome (40% dos casos), além da matriarca. Exoforia (para perto e para longe) foi identificada em 3 casos (30%). Os outros 3 pacientes não apresentavam alterações da motilidade extrínseca ocular.
Todos os pacientes apresentaram pressão ocular (PO) abaixo de 18 mmHg e nenhum disco óptico foi identificado como suspeito de glaucoma.
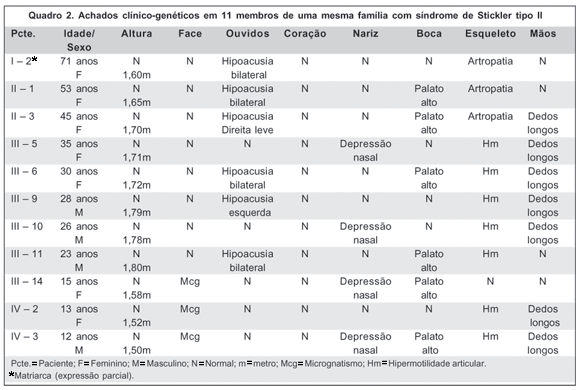
O exame clínico-genético revelou que 30% dos pacientes com SS tipo II apresentavam micrognatia, 50% (5 pacientes) com hipoacusia, 40% (4 pacientes) com depressão nasal e 60% (6 pacientes) apresentavam palato alto. Hipermotilidade articular e dedos longos foram demonstrados em 7 casos (70%), sendo que 6 pacientes (60%) apresentavam hipermotilidade articular juntamente com dedos longos. Artropatia esteve presente em 3 pacientes (30% dos casos) (Quadro 2).
DISCUSSÃO
A síndrome de Stickler (artro-oftalmopatia hereditária) é uma vitreo-retinopatia e condrodisplasia que pode causar alterações sistêmicas e oftalmológicas importantes. Porém, a dificuldade no diagnóstico diferencial com outras alterações genéticas como a síndrome de Marshall ou fenótipo Marshall/Stickler(21), síndrome de Wagner(20,23), alta miopia familial(26), subestima o número de pacientes com síndrome de Stickler.
A alta miopia desde a infância e as alterações vítreo-retinianas são típicas da SS tipo II e foram encontradas na maioria dos pacientes deste estudo. Entretanto, a subluxação de cristalino, que também esteve presente em vários pacientes, não é citada na literatura como fator importante na distinção diagnóstica. Esses pacientes são confundidos, muitas vezes, com a síndrome de Marfan, na qual o cristalino também se encontra subluxado(27). A distinção se fez em relação ao local da subluxação, súpero-lateralmente na maioria dos pacientes com síndrome de Marfan e súpero-medialmente e bilateral em todos os pacientes com subluxação avaliados neste artigo, além de não haver nenhum paciente com comprometimento cardíaco ou qualquer outra alteração típica da síndrome de Marfan, como por exemplo, aracnodactilia (em 88% dos casos)(27). Para complementar a distinção sindrômica, o PPMF indicou alteração marfanóide, entretanto diferente das alterações encontradas na síndrome de Marfan(25).
A baixa de acuidade visual se deve, não somente à alta miopia, mas sim às inúmeras alterações vítreo-retinianas e cristalineanas. Catarata, glaucoma e DR são freqüentemente citados na literatura, porém neste trabalho não se evidenciou caso de glaucoma e somente um caso de DR foi diagnosticado(1,16-17). Já em relação à catarata, é fundamental a boa correlação com a patologia em si e não subentender como catarata os pacientes com SS submetidos à facoemulsificação por outras causas como: subluxação, luxação ou iridodonese. Pseudofacia com implante de anel de tensão foi identificada em 2 pacientes neste estudo, porém os pacientes não sabiam relatar a indicação cirúrgica (se foi realizada somente pela alteração do posicionamento do cristalino ou se havia opacidade cristalineana conjuntamente).
Toda miopia em criança deve ser acompanhada com muito cuidado, sempre pesquisando alterações sistêmicas, já que muitas são as síndromes genéticas que cursam com miopia congênita, dentre elas: síndrome de Down, síndrome de Marfan, homocistinúria, síndrome de Ehlers-Danlos, síndrome de Stickler, síndrome de Pierre-Robin, síndrome de Prader-Willi, síndrome de Noonan entre outras(26,28). A miopia presente na SS tipo II é descrita normalmente como não progressiva. Entretanto, não podemos deixar de acompanhar rigorosamente os portadores de SS devido a possível associação com outras síndromes e alterações, como a alta miopia hereditária. A possibilidade de uso do laser para degenerações miópicas não deve ser descartada.
Sendo uma anomalia do colágeno, a SS tipo II afeta principalmente a arquitetura do vítreo, e esse é um importante marcador da síndrome(7). Não obstante, devido à vasta combinação genética e aos inúmeros mecanismos de inter-relação gênica, as várias facetas de uma síndrome genética podem ser mimetizadas. Alterações vítreo-retinianas são importantes na distinção sindrômica, mas o conjunto de informações clínicas assume papel de importância ainda maior. Os 20% de pacientes com SS tipo II que não apresentaram deformidades vítreo-retinianas demonstram a expressão parcial e alta variabilidade fenotípica da síndrome. Há necessidade de exames laboratoriais para abordagem gênica e constatação de prováveis mutações, alterações da herança ou até mesmo uma provável ligação entre SS tipo I e tipo II. A correlação entre os vários tipos de colágeno já é bastante conhecida e pode explicar a difícil tarefa na distinção das diversas modalidades genéticas, mesmo que somente mutações no gene COL11A1 (atuação no colágeno XI), possam provocar SS tipo II(24). Essa grande variabilidade fenotípica também foi relatada em outro trabalho que comprovou defeito no colágeno tipo XI (COL11A1) em 3 pacientes com síndrome de Marshall (mesmo alelo da SS tipo II), porém os achados clínico-genéticos provaram que a síndrome de Marshall e a síndrome de Stickler são distintas(22). A síndrome de Marshall é caracterizada por estatura baixa, miopia, catarata congênita, esotropia, luxação do cristalino, degeneração vítrea, perda auditiva, e outras alterações que podem confundir com a SS tipo II(21-22). Neste artigo, no entanto, não houve pacientes com altura baixa, todos os pacientes com alteração da motilidade não apresentavam esotropia e sim alterações divergentes, além das alterações vítreas serem típicas da SS tipo II.
Atualmente discute-se sobre a real alteração do colágeno presente no vítreo. Através da análise ultra-estrutural do colágeno foi demonstrada a presença de fenestrações na membrana vítrea de uma paciente com SS tipo II, porém as fibras de colágeno eram similares às do vítreo normal. Este estudo sugere que, provavelmente, não há alteração no colágeno e sim, quantidade insuficiente de colágeno durante o desenvolvimento vítreo(29).
Outros achados foram associados com a SS tipo II, dentre eles: degeneração radial perivascular da retina(16), atrofia corrioretiniana posterior(17) e DR reugmatogênico pediátrico(30). Recentemente foi relatado um caso de morte por introdução de cateter cirúrgico oftalmológico na fissura orbitária superior, comprovando a fragilidade do tecido conectivo orbitário em pacientes com SS(31). Devido a essas alterações, pacientes com SS submetidos a intervenção cirúrgica podem apresentar complicações mais freqüentes, principalmente no que diz respeito à fragilidade zonular, nas cirurgias de catarata, fragilidade escleral e vítreo-retiniana, nas cirurgias de estrabismo ou de retina e vítreo.
Alterações da motilidade extrínseca ocular não são citadas com freqüência pela literatura em pacientes com SS tipo II, porém neste trabalho há grande predomínio do exodesvio. No entanto, como a SS tipo II, o estrabismo também apresenta alterações genéticas ainda obscuras e a sobreposição de alterações de cunho gênico não é improvável.
Em alguns casos foram pedidos exames de ultra-sonografia ocular devido à dificuldade de visualização correta da retina, porém não houve retorno da maioria dos pacientes ao hospital.
As alterações orofaciais (principalmente a micrognatia) estiveram presentes nas crianças e a artropatia, nos pacientes com mais idade, sendo esses achados compatíveis com a literatura que cita as alterações orofaciais como mais evidentes na infância e de diagnóstico difícil com o aumento da idade(19,32). Neste artigo a hipermotilidade articular e a presença de dedos longos foram diagnosticados entre pacientes jovens, ao contrário das artropatias, presentes somente nos pacientes com mais de 44 anos, o que reforça os dados da literatura que relatam hipermotilidade em muitos pacientes jovens e desenvolvimento de artropatia em pessoas na terceira ou quarta década de vida(19,33).
Em relação a essa família em estudo, a perda auditiva esteve presente em 5 pacientes (50% dos casos), sendo também compatível com os dados da literatura que indicam em torno de 40% de hipoacusia em pessoas com SS tipo II(19). São duas as possíveis razões para a perda auditiva: presença de palato alto aumentando a incidência de otite média levando a alterações dos ossículos auditivos ou simplesmente por se tratar de uma alteração genética associada(32). Neste trabalho houve presença de palato alto em 4 dos 5 pacientes com hipoacusia que apresentavam SS tipo II.
CONCLUSÃO
A confrontação dos dados clínico-genéticos com os achados oculares não revelou nenhuma associação de grande importância. A presença de uma alteração ocular não pode ser associada a um dado clínico-genético específico. Há grande variação fenotípica, ocular ou sistêmica, entre os pacientes examinados. Os sinais e sintomas devem ser considerados em conjunto.
Os achados encontrados, no entanto, são de indivíduos de uma mesma família, fato que influencia na generalização dos dados encontrados.
A distinção sindrômica, o acompanhamento precoce, freqüente e multidisciplinar, além do tratamento e aconselhamento genético, são condutas médicas importantes para estes indivíduos.
REFERÊNCIAS
1. Stickler GB, Belau PG, Farrell FJ, Jones JD, Pugh DG, Steinberg AG, et al. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc. 1965;40:433-55.
2. Francomano CA, Liberfarb RM, Hirose T, Maumenee IH, Streeten EA, Meyers DA, et al. The Stickler syndrome: evidence for close linkage to the structural gene for type II collagen. Genomics. 1987;1(4):293-6.
3. Knowlton RG, Weaver EJ, Struyk AF, Knobloch WH, King RA, Norris K, et al. Genetic linkage analysis of hereditary arthro-ophthalmopathy (Stickler syndrome) and the type II procollagen gene. Am J Hum Genet. 1989;45(5):681-8.
4. Zlotogora J, Sagi M, Schuper A, Leiba H, Merin S. Variability of Stickler syndrome. Am J Med Genet. 1992;42(3):337-9.
5. Liberfarb RM, Levy HP, Peter SR, Wilkin DJ, Davis J, Balog JZ, et al. The Stickler syndrome: genotype/phenotype correlation in 10 families with Stickler syndrome resulting from 7 mutations in the type II collagen gene locus COL2A1. Genet Med. 2003;5(1):21-7. Erratum in: Genet Med. 2003; 5(6):478.
6. Sirko-Osadsa DA, Zlotogora J, Tiller GE, Knowlton RG, Warman ML. A third Stickler syndrome locus is linked to COL11A1, the gene encoding the alpha-1 subunit of collagen XI. Am J Hum Genet. 1996;59(Suppl.):A17.
7. Richards AJ, Yates JR, Williams R, Payne SJ, Pope FM, Scott JD, et al. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha-1(XI) collagen. Hum Molec Genet. 1996;5(9):1339-43.
8. Martin S, Richards AJ, Yates JRW, Scott JD, Pope FM, Snead MP. Stickler syndrome: further mutations in COL11A1 and evidence for additional locus heterogeneity. Eur J Hum Genet. 1999;7(7):807-14.
9. Brunner HG, Van Beersum SE, Warman ML, Olsen BR, Ropers HH, Mariman EC. A Stickler syndrome gene is linked to chromosome 6 near the COL11A2 gene. Hum Molec Genet. 1994;3(9):1561-4.
10. Vikkula M, Mariman EC, Lui VCH, Zhidkova NI, Tiller GE, Goldring MB, et al. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell. 1995;80(3):431-7. Comment in: Cell. 1995; 80(3):371-8.
11. Ahmad N, Richards AJ, Murfett HC, Shapiro L, Scott JD, Yates JRW, et al. Prevalence of mitral valve prolapse in Stickler syndrome. Am J Med Genet 2003;116(3):234-7.
12. Stickler GB, Hughes W, Houchin P. Clinical features of hereditary progressive arthro-ophthalmopathy (Stickler syndrome): a survey. Genet Med. 2001;3(3): 192-6.
13. Regenbogen L, Godel V. Hereditary degeneration, cheft lip and palate, deafness, and skeletal dysplasia. Am J Ophthal. 1980;89(3):414-8.
14. Masmoudi S, Tlili A, Majava M, Ghorbel AM, Chardenoux S, Lemainque A, et al. Mapping of a new autosomal recessive nonsyndromic hearing loss locus (DFNB32) to chromosome 1p13.3-22.1. Eur J Hum Genet. 2003;11(2): 185-8.
15. Donoso LA, Edwards AO, Frost AT, Ritter R, Ahmad N, Vrabec T, et al. Clinical variability of Stickler syndrome: role of exon 2 of the collagen COL2A1 gene. Surv Ophthalmol. 2003;48(2):191-203.
16. Parma ES, Körkkö J, Hagler WS, Ala-Kokko L. Radial perivascular retinal degeneration: a key to the clinical diagnosis of an ocular variant of Stickler syndrome with minimal or no systemic manifestations. Am J Ophthalmol. 2002;134(5):728-34. Comment in: Am J Ophthalmol. 2002;134(5):746-8.
17. Vu CD, Brown J Jr, Korkko J, Ritter R 3rd, Edwards AO. Posterior chorioretinal atrophy and vitreous phenotype in a family with Stickler syndrome from a mutation in the COL2A1 gene. Ophthalmology. 2003;110(1):70-7.
18. Sarra GM, Weigell-Weber M, Kotzot D, Neimeyer G, Messmer E, Hergersberg M. Clinical description and exclusion of candidate genes in a novel autosomal recessively inherited vitreoretinal dystrophy. Arch Ophthalmol. 2003;121(8): 1109-16.
19. Snead MP, Yates JRW. Clinical and molecular genetics of Stickler syndrome. J Med Genet. 1999;36(5):353-9.
20. Griffith AJ, Sprunger LK, Sirko-Osadsa DA, Tiller GE, Meisler MH, Warman ML. Marshall syndrome associated with a splicing defect at the COL11A1 gene. Am J Hum Genet. 1998;62(4):816-23.
21. Annunen S, Korkko J, Czarny M, Warman ML, Brunner HG, Kaariainen H, et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999;65(4):974-83.
22. Gonçalves Jr A. Síndrome de Marshal: síndrome distinta ou uma variante da Síndrome de Stickler? Rev Bras Oftalmol. 1999;58(6):465-71.
23. Richards AJ, Martin S, Yates JRW, Scott JD, Baguley DM, Pope FM, et al. COL2A1 exon 2 mutations: relevance to the Stickler and Wagner syndromes. Br J Ophthalmol. 2000;84(4):364-71. Comment in: Am J Ophthalmol. 2002; 134(3):473; author reply 473-4.
24. Richards AJ, Baguley DM, Yates JR, Lane C, Nicol M, Harper PS, et al. Variation in the vitreous phenotype of Stickler syndrome can be caused by different amino acid substitutions in the X position of the Type II collagen Gly-X-Y triple helix. Am J Hum Genet. 2000;67(5):1083-94.
25. de Oliveira Sobrinho RP, Moretti-Ferreira D, Contini A, Norato DY. Metacarpophalangeal pattern profile in Marfan syndrome and Marfan-like patients. Am J Med Genet. 1997;72(2):159-63.
26. Young TL, Ronan SM, Drahozal LA, Wildenberg SC, Alvear AB, Oetting WS, et al. Evidence that a locus for familial high myopia maps to chromossome 18p. Am J Hum Genet. 1998;63(1):109-19.
27. Ghanem VC, Ghanem EA, Ghanem RC, Arieta CEL. Monoscleral fixation of IOL after extracapsular extraction of subluxated lenses in patients with Marfan syndrome. Arq Bras Oftalmol. 2004;67(5):763-7.
28. Cunha RNP. Miopia na infância. Arq Bras Oftalmol. 2000;63(3):231-4.
29. Betis F, Hofman P, Gastaud P. Modifications vitréennes au cours d'un syndrome de Stickler. J Fr Ophthalmol. 2003;26(4):386-90.
30. Fivgas GD, Capone A. Pediatric rhegmatogenous retinal detachment. Retina. 2001;21(2):101-6.
31. Grag S, Piva A, Sanchez RN, Sadun AA. Death associated with an indwelling orbital catheter. Opththal Plast Reconstr Surg. 2003;19(5):398-400.
32. Temple IK. Stickler's syndrome. J Med Genet. 1989;26(2):119-26.
33. Rai A, Wordsworth P, Coppock JS, Zaphiropoulos GC, Struthers GR. Hereditary arthro-ophthalmopathy (Stickler syndrome): a diagnosis to consider in familial premature osteoarthritis. Br J Rheumatol. 1994;33(12):1175-80.
Endereço para correspondência:
Vanderson Glerian Dias
Rua Guaporé, 432
Catanduva (SP) CEP 15804-105
E-mail: [email protected]
Recebido para publicação em 11.05.2005
Última versão recebida em 02.06.2006
Aprovação em 05.06.2006
Trabalho realizado pelo Departamento de Oftalmologia do Hospital Universitário da Universidade do Oeste Paulista - Presidente Prudente (SP) - Brasil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Solange Rios Salomão sobre a divulgação de seu nome como revisor, agradecemos sua participação neste processo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.