Arq. Bras. Oftalmol. 2004; 67 (6): 10.1590/S0004-27492004000600023
Total: 3638
Bruno Machado Fontes1; Jane Chen1; André Hasler Príncipe1; Juliana M. Ferraz Sallum2; Carlos Filipe Chicani3
DOI: 10.1590/S0004-27492004000600023
RESUMO
A síndrome de Wolfram consiste na associação de diabetes mellitus e atrofia óptica. Outros achados comuns são surdez neurossensorial, alterações do trato urinário e distúrbios neurológicos. Tem padrão de herança autossômico recessivo com penetrância incompleta e expressividade variável. O objetivo deste relato é apresentar paciente que apresenta todas as características da síndrome de Wolfram (ou síndrome DIDMOAD). JFP, negro, 23 anos, apresenta diabetes mellitus e insipidus, atrofia óptica, surdez neurossensorial, polineuropatia periférica, neuropatia autonômica, bexiga neurogênica, dilatação do trato urinário, infecções repetidas do trato urinário e azoospermia. Os exames clínico-oftalmológico, retinografia, angiografia fluoresceínica, eletrorretinografia (ERG) e potencial visual evocado (PVE) mostram padrão de normalidade retiniana e de atrofia de nervos ópticos. A síndrome de Wolfram deve ser lembrada em casos de atrofia óptica associados a diabetes, poliúria, polidipsia ou a qualquer uma das alterações apresentadas.
Descritores: Síndrome de Wolfram; Atrofia óptica; Diabetes mellitus; Diabetes insípido; Surdez
ABSTRACT
Wolfram syndrome consists of the association of diabetes mellitus with optic atrophy. Other common findings are deafness, urinary tract and neurological disorders. It is an autossomic recessive disease, with incomplete penetrance and variable expressivity. The aim of this case report is to describe a patient who presents all the characteristics of Wolfram syndrome (DIDMOAD syndrome). JFP, African - American, 23 years old, presents with diabetes mellitus and insipidus, optic atrophy, deafness, peripheral polyneuropathy, autonomic neuropathy, neurogenic bladder, urinary tract dilation with recurrent infections, and azoospermia. Clinical examination, retinography, fluorescein angiogram, eletroretinography (ERG) and visual evocated potencial (VEP) revealed no retinal disorders and bilateral optic atrophy. Wolfram syndrome must be remembered in cases of optic atrophy in association with diabetes, urinary disorders, or any of the described alterations.
Keywords: Wolfram Syndrome; Optic atrophy; Diabetes mellitus; Diabetes insipidus; Deafness
INTRODUÇÃO
A síndrome descrita em 1938 por Wolfram(1), batizada posteriormente com seu nome, contava com a presença da associação de diabetes mellitus e atrofia óptica em uma mesma família. Apresentada posteriormente por Pilley e Thompson(2) com o termo DIDMOAD (Diabetes Insipidus, Diabetes Mellitus Optic Atrophy e Deafness), caracteriza-se por ser uma doença onde estão presentes o diabetes insipidus (73%), diabetes mellitus (100%), atrofia óptica (100%) e surdez (62%)(3-4). Outras alterações oculares e sistêmicas podem encontrar-se associadas, como catarata, retinopatia diabética, glaucoma, alterações do trato urinário (58%), dismotilidade gastrintestinal (24%), distúrbios neurológicos (62%), psiquiátricos e atrofia gonadal(3-11).
Pouco mais de trezentos casos foram descritos na literatura mundial, sendo a maioria com a síndrome incompleta. Nosso objetivo com o presente relato é o de apresentar um caso com todos os achados clássicos da síndrome.
CASO CLÍNICO
Paciente de 23 anos, sexo masculino, negro, compareceu ao ambulatório de Neuro-Oftalmologia da UNIFESP com queixa de baixa de acuidade visual progressiva, com início aos 12 de idade, acompanhada de fotofobia. Teve o diagnóstico de diabetes insipidus e mellitus insulino-dependente aos 3 anos de idade, quando apresentou quadro de poliúria, polidipsia e perda ponderal. Apresentava ainda hipoacusia severa e progressiva (uso de prótese auditiva desde os 14 anos de idade), polineuropatia periférica, bexiga neurogênica e dilatação do trato urinário complicadas com infecções urinárias de repetição. Espermograma mostrava azoospermia, hemospermia e leucospermia.
Mãe com hipertensão arterial sistêmica e diabetes mellitus não insulino-dependente, ambos controlados e com diagnóstico na idade adulta. Nenhum outro caso de doença sistêmica ou ocular na família.
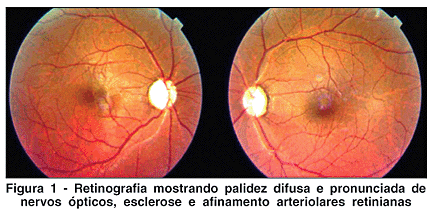
O exame oftalmológico inicial mostrou desvio ocular (esotropia) do olho direito, pupilas em meia midríase, anisocóricas (direita maior que esquerda) e não reagentes ao estímulo luminoso ou à acomodação. A acuidade visual medida pela tabela ETDRS sem correção foi de conta dedos a 50 cm em olho direito e 20/800 em olho esquerdo, não obtendo melhora com buraco estenopeico. A biomicroscopia não revelou anormalidades além das relatadas à pupila, e a pressão intra-ocular, medida com tonômetro de aplanação, mostrou-se dentro dos padrões de normalidade (16 mmHg em ambos os olhos). Motilidade ocular normal. Apresentava na fundoscopia uma palidez de nervo óptico difusa pronunciada, com escavação fisiológica em ambos os olhos, além de um aumento do reflexo dorsal das artérias retinianas (Figura 1).
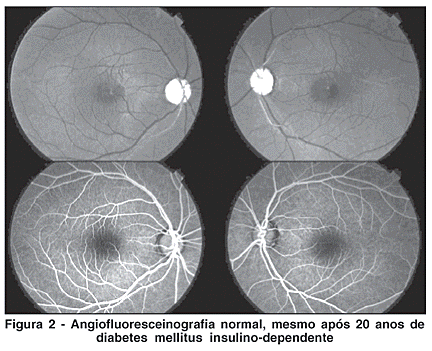
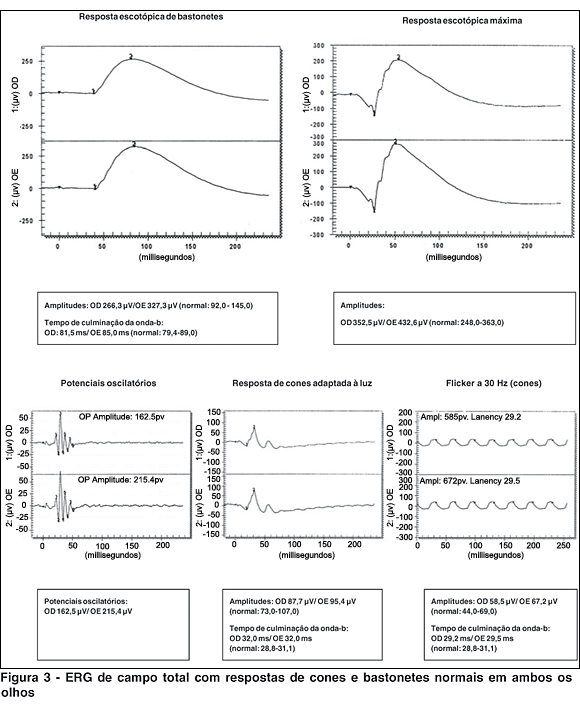
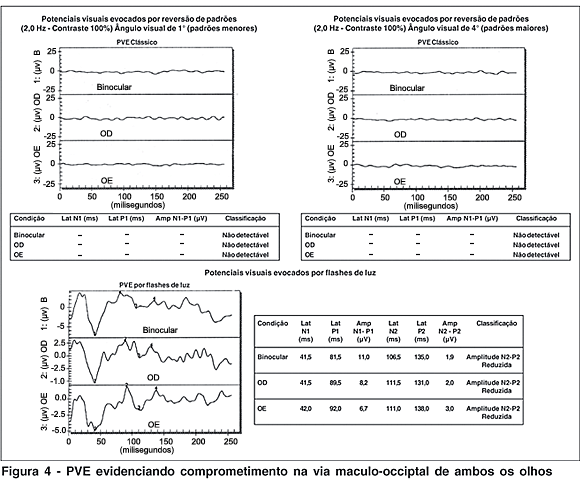
A angiografia fluoresceínica não mostrou sinais de retinopatia diabética, mesmo após 20 anos de doença de difícil controle (Figura 2). A eletrorretinografia de campo total apresentou respostas de cones e bastonetes normais em ambos os olhos (Figura 3). Os registros dos potenciais visuais evocados evidenciaram comprometimento severo na via maculo- occiptal de ambos os olhos, compatível com envolvimento bilateral dos nervos ópticos (Figura 4). Não foram realizados os exames de campo visual e de visão de cores devido à baixa acuidade visual.
DISCUSSÃO
A síndrome de Wolfram é uma entidade rara. Barret et al(4) reportaram uma prevalência de 1 caso para 770.000 pessoas na Inglaterra. Fraser & Gunn(12) estimaram sua prevalência em 1 a cada 100.000 crianças, baseados na freqüência de atrofia óptica em uma clínica de diabetes juvenil de 1/148.
Uma disfunção mitocondrial [mutação do tRNA Leu (3243)] tem sido implicada como causa desta doença, que tem padrão autossômico recessivo de herança com penetrância incompleta e expressividade variável(3-4,7,9,11,13-17). O gene responsável é o WFS1 (OMIM 606201), membro de uma família que codifica 890 glicoproteínas de aminoácidos, que contém domínios C - hidrofílicos e N - terminais, com 10 domínios transmembrana hidrofóbicos(11-16). Está presente no cromossomo 4p16.3 e consiste de 8 exons(9,11-16). Mutações neste gene levam à morte das células beta pancreáticas, sendo diretamente responsáveis pelo diabetes mellitus nestes pacien tes(16). Um outro locus descrito para a doença foi mapeado em 4q (WFS2; OMIM 604928).
Entre os diagnósticos diferenciais temos a síndrome da rubéola congênita, atrofia óptica hereditária de Leber e anemia "thiamine-responsive" com diabetes mellitus e surdez.
Além da atrofia óptica (presente em todos os casos), outras alterações oculares são descritas na literatura, tais como catarata(3,8-10), distúrbios da visão de cor(3-4,10), anormalidades pupilares(3,5-6,10), miopia(5,9), alterações do epitélio pigmentar da retina(6,10), nistagmo(6,10), perda de campo visual(4-7,9) e glaucoma(9).
Um dado interessante baseado na literatura é a pequena freqüência de retinopatia diabética encontrado, apesar destes pacientes apresentarem diabetes mellitus com difícil controle glicêmico por um longo tempo(3-10).
REFERÊNCIAS
1. Wolfram DJ. Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Proc Mayo Clin 1938;13:715-8.
2. Pilley SF, Thompson HS. Familial syndrome of diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (didmoad) in childhood. Br J Ophthalmol. 1976;60(4):294-8.
3. Reis AF, Ferreira JG, Coifman R, Russo EMK, Moisés RCS, Dib SA. Síndrome de Wolfram: relato de um caso, revisão da literatura e caracterização do diabetes mellitus associado. Arq Bras Endocrinol Metab. 1996;40(2):106-12.
4. Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet. 1995;346 (8988):1458-63.
5. Pacini Neto L, Silva JA, Garcia O. Síndrome de Wolfram: relato de três casos. Rev Bras Oftalmol. 1993;52(5):49-53.
6. Leite EM, Oliveira RBC. Acompanhamento oftalmológico a longo prazo da síndrome de Wolfram. Rev Bras Oftalmol. 1996;55(3):49-55.
7. Van den Bergh L, Zeyen T, Verhelst J, Mahler C. Wolfram syndrome: a clinical study of two cases. Doc Ophthalmol. 1993; 84(2):119-26.
8. Castro FJ, Barrio J, Perena MF, Palomar MT, Cristóbal JA. Uncommon ophthalmologic findings associated with Wolfram syndrome. Acta Ophthalmol Scand. 2000;78(1):118-9.
9. Bekir NA, Gungor K, Guran S. A DIDMOAD syndrome family with juvenile glaucoma and myopia findings. Acta Ophthalmol Scand. 2000;78(4):480-2.
10. Al-Till M, Jarrah NS, Ajlouni KM. Ophthalmologic findings in fifteen patients with Wolfram syndrome. Eur J Ophthalmol. 2002;12(2):84-8.
11. Crawford J, Zielinski MA, Fisher LJ, Sutherland GR, Goldney RD. Is there a relationship between Wolfram syndrome carrier status and suicide ? Am J Med Genet. 2002;114(3):343-6.
12. Fraser FC, Gunn T. Diabetes mellitus, diabetes insipidus, and optic atrophy. An autosomal recessive syndrome ? J Med Genet. 1977;14(3):190-3.
13. Seyrantepe V, Topaloglu H, Simsek E, Ozguc M, Yordam N. Mitochondrial DNA studies in Wolfram (DIDMOAD) syndrome. Lancet. 1996;347(9002):695-6.
14. Bespalova IN, Van Camp G, Bom SJ, Brown DJ, Cryns K, DeWan AT, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet. 200;10(22):2501-8.
15. Young TL, Ives E, Lynch E, Person R, Snook S, MacLaren L, et al. Non - syndromic progressive hearing loss DFNA 38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet. 2001;10(22):2509-14.
16. Minton JA, Hattersley AT, Owen K, McCarthy MI, Walker M, Latif F, et al. Association studies of genetic variation in the WFS1 gene and type 2 diabetes in U.K. populations. Diabetes.2002;51(4):1287-90.
17. Sallum JMF, Farah MEF, Maumenee IH. Heterogeneidade genética em atrofia óptica autossômica dominante. Arq Bras Oftalmol. 2002;65(4):419-26.
Endereço para correspondência
Bruno Machado Fontes
Rua Pedro de Toledo 544 / 403
São Paulo (SP) CEP 04023-000
E-mail: [email protected]
Recebido para publicação em 04.02.2004
Versão revisada recebida em 15.07.2004
Aprovação em 22.07.2004
Trabalho realizado nos Setores de Retina e Vítreo e Neuro-Oftalmologia do Departamento de Oftalmologia da Universidade Federal de São Paulo - UNIFESP.
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos ao Dr. Mário Luiz Ribeiro Monteiro.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.