Arq. Bras. Oftalmol. 2013; 76 (6): 10.1590/S0004-27492013000600015
Total: 2425
Daniela Fernandes de Carvalho Rios1; Luis Felipe da Silva Alves Carneiro1; Aline Amaral Fulgêncio da Cunha1; Carlos Bernardo Moura Dalle1; Maria Frasson2
DOI: 10.1590/S0004-27492013000600015
ABSTRACT
Unilateral retinitis pigmentosa is a rare disease characterized by loss of photoreceptors and retinal pigment deposition without affecting the contralateral eye. Although described more than one hundred years its existence is still questioned. This article reports a case of a patient with abnormalities suggestive of unilateral retinitis pigmentosa. The clinical and complementary examinations are discussed.
Keywords: Retinitis pigmentosa; Retinal diseases; Electroretinography; Fluorescein angiography; Humans; Male; Adult; Case report
RESUMO
A retinose pigmentar unilateral é uma doença rara caracterizada pela perda de fotorreceptores e deposição de pigmento na retina sem acometimento do olho contralateral. Apesar de descrita há mais de cem anos sua existência ainda é questionada. Este artigo relata o caso de um paciente com alterações sugestivas de retinose pigmentar unilateral. Os aspectos clínicos e os exames complementares são discutidos.
Descritores: Retinite pigmentosa; Doenças retinianas; Eletrorretinografia; Angiofluoresceinografia; Humanos; Masculino; Adulto; Relato de caso
RELATO DE CASO
Retinose pigmentar unilateral ou pseudorretinose pigmentar?: relato de caso
Unilateral retinitis pigmentosa or pseudoretinitis pigmentosa?: case report
Daniela Fernandes de Carvalho RiosI; Luis Felipe da Silva Alves CarneiroI; Aline Amaral Fulgêncio da CunhaI; Carlos Bernardo Moura DalleI; Maria FrassonII
IMédico, Clínica de Olhos do Hospital Santa Casa de Belo Horizonte, Belo Horizonte (MG), Brasil
IIMédica, Departamento de Oftalmologia, Hospital das Clínicas da Universidade Federal de Minas Gerais, Belo Horizonte (MG), Brasil
RESUMO
A retinose pigmentar unilateral é uma doença rara caracterizada pela perda de fotorreceptores e deposição de pigmento na retina sem acometimento do olho contralateral. Apesar de descrita há mais de cem anos sua existência ainda é questionada. Este artigo relata o caso de um paciente com alterações sugestivas de retinose pigmentar unilateral. Os aspectos clínicos e os exames complementares são discutidos.
Descritores: Retinite pigmentosa/diagnóstico; Doenças retinianas; Eletrorretinografia; Angiofluoresceinografia; Humanos; Masculino; Adulto; Relato de caso
ABSTRACT
Unilateral retinitis pigmentosa is a rare disease characterized by loss of photoreceptors and retinal pigment deposition without affecting the contralateral eye. Although described more than one hundred years its existence is still questioned. This article reports a case of a patient with abnormalities suggestive of unilateral retinitis pigmentosa. The clinical and complementary examinations are discussed.
Keywords: Retinitis pigmentosa/diagnosis; Retinal diseases; Electroretinography; Fluorescein angiography; Humans; Male; Adult; Case report
INTRODUÇÃO
A retinose pigmentar é um termo utilizado para um grupo de doenças que são caracterizadas pela hereditariedade, bilateralidade, disfunção progressiva, perda celular e eventual atrofia retiniana. A perda dos fotorreceptores leva a posterior deterioração das camadas internas da retina, com atrofia difusa de todas as camadas. Os sinais característicos são: aglomerados pigmentares com aspecto de "espículas ósseas" na periferia da retina, atenuação dos vasos e palidez "cérea" do nervo óptico. As primeiras descrições de retinose pigmentar unilateral (RPU) datam de meados do século XIX(1). Desde então, vários outros foram relatados. Em 1948, Dreisler sugeriu que essas condições representavam uma rara forma de manifestação assimétrica da doença bilateral(2) e em 1951, Joseph concluiu que muitos destes eram secundários a outras doenças(3).
Ainda hoje persiste a discussão sobre a existência da RPU como entidade clínica isolada, sua relação com a retinose pigmentar bilateral (RPB) e, principalmente, sua associação com outras doenças adquiridas. Estas incertezas podem confundir o médico oftalmologista especialmente quando não é encontrada uma causa para as alterações apresentadas ao exame clínico.
RELATO DE CASO
Paciente do sexo masculino, 24 anos, procedente de Belo Horizonte foi admitido na unidade de urgência da Clínica de Olhos do Hospital Santa Casa de Belo Horizonte, com relato de diminuição da acuidade visual no olho direito (OD) com evolução de dois dias. Negou dor, trauma ocular recente ou prévio. Informou passado de varicela aos 19 anos. Sua história familiar evidenciou mãe com hipertensão arterial sistêmica. Ao exame oftalmológico apresentou acuidade visual corrigida de 0,4 (20/50) no OD e 1,0 (20/20) no olho esquerdo (OE). A biomicroscopia do segmento anterior não revelou alterações e a medida da pressão intraocular pela tonometria de aplanação (Goldmann) foi de 16 mmHg em ambos os olhos. O mapeamento de retina do OD evidenciou vítreo claro, retina aplicada, atenuação arteriolar difusa, disco óptico corado com contornos regulares e relação escavação/disco fisiológica, rarefação de epitélio pigmentar da retina (EPR) e aglomerados pigmentares similares à "espículas ósseas" 3600 poupando a região macular. O exame do OE demonstrou vítreo claro, retina aplicada, vasos retinianos com relação artéria/veia de 2/3, disco óptico corado, com contornos regulares e relação escavação/disco fisiológica, brilho macular preservado sem qualquer alteração retiniana.
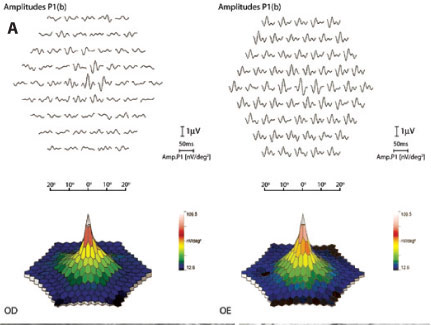
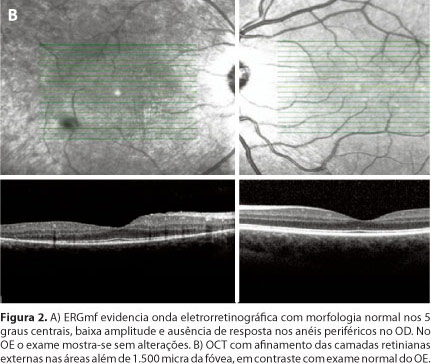
O campo visual manual (CVM) realizado com o campímetro manual (Goldmann) mostrou padrão tubular no OD e exame sem alterações no OE (Figura 1A). A retinografia colorida utilizando o equipamento Spectralis (Heidelberg Engineering) evidenciou atenuação dos vasos e aglomerados pigmentares além das arcadas vasculares no OD, não observadas no OE (Figura 1B). Angiografia fluorescente realizada com o mesmo aparelho demonstrou áreas de hipofluorescência por efeito máscara devido aos acúmulos pigmentares, hiperfluorescência por efeito janela em correspondência à ausência de pigmento no EPR além das arcadas vasculares no OD. Na avaliação da autofluorescência observou-se padrão numular hipofluorescente na média periferia no OD e dentro dos padrões da normalidade no OE (Figura 1C). O eletrorretinograma (ERG) de campo total utilizando o sistema Retiport/Retiscan (Roland Consult) e eletrodo tipo ERG Jet mostrou respostas extintas no OD tanto nas fases escotópicas quanto nas fases fotópicas. No OE a amplitude e o tempo implícito das ondas a e b foram dentro dos padrões da normalidade. O ERG multifocal (ERGmf) realizado com o aparelho Retscan System (Roland Consult) demonstrou onda eletrorretinográfica com morfologia normal nos 5 graus centrais, apesar de baixa amplitude e ausência de resposta nos anéis periféricos. No OE o exame mostrou-se sem alterações (Figura 2A). A tomografia de coerência óptica (OCT) com o equipamento Spectralis (Heidelberg Engineering) evidenciou afinamento das camadas retinianas externas nas áreas além de 1.500 micra da fóvea (espessura de aproximadamente 30 micra), sugerindo acometimento dos fotorreceptores no OD, em contraste com exame normal (aproximadamente 88 micra) no OE (Figura 2B).
Os exames complementares sistêmicos evidenciaram: "Venereal Disease Research Laboratory test" (VDRL) e teste de absorção do anticorpo treponêmico fluorescente (FTA-ABS) negativos, sorologia para toxoplasmose IgM negativo e IgG positivo, hemograma e velocidade de hemossedimentação (VHS) sem alterações. A investigação oftalmológica de seus familiares não evidenciou alterações similares às do paciente. O mesmo foi acompanhado periodicamente e manteve as alterações já descritas.
DISCUSSÃO
A RPU tem sido descrita na literatura, mas ainda existem dúvidas quanto à existência de uma entidade clínica isolada e sua relação com a forma bilateral(3). Na RPB os casos hereditários representaram 34% em um estudo, porcentagem similar ao observado na doença unilateral (36%) falta referência. Praticamente todas as formas de hereditariedade mendeliana e não mendeliana foram descritas na RPB. Segundo alguns trabalhos a RPU também apresentaria uma hereditariedade semelhante, mas não foram identificados casos relacionados ao cromossoma X(4). Mais de 45 genes foram identificados para RPB sendo que estes correspondem a apenas 60% dos pacientes com a doença, o restante destes pacientes possui, possivelmente, alterações em genes ainda desconhecidos o que dificulta sua confirmação genética(5).
Muitas vezes a RPB pode ser assimétrica e o acometimento no olho, em princípio, sadio ocorrer muitos anos após a manifestação inicial da doença. Contudo, nesses casos, o ERG já apresenta alterações em ambos os olhos desde o início da doença(6). O tempo ideal de acompanhamento do paciente para o diagnostico de RPU ainda é controverso. Alguns autores sugerem um período mínimo de 5 anos(7) mas há relatos de pacientes que desenvolveram a doença bilateral após 10 anos(8).
O diagnóstico da RPU pode ser tardio porque muitas vezes o paciente apresenta visão preservada em um olho e somente percebe a baixa acuidade visual quando esta é grave(4).
Em relação aos sinais e sintomas, assim como as alterações de exames complementares não há diferenças entre as duas doenças. François, Verriest sugeriram critérios para o diagnóstico da RPU: excluir todas as etiologias infecciosas e checar que os sinais clínicos da retinose estão presentes em um olho e totalmente ausentes no outro olho(7).
Outras retinopatias pigmentares chamadas de pseudorretinoses podem simular RPU como, por exemplo, doenças infecciosas e inflamatórias prévias da retina (sífilis, toxoplasmose, rubéola, tuberculose e outras viroses). Doenças como o melanoma de coroide(9), angioma de coroide(10) e lupus eritematoso sistêmico(11) também foram associadas à pseudorretinose pigmentar.
No caso em discussão, foram descartadas algumas causas infecciosas, assim como traumas (descolamento prévio de retina) e uso de medicações (retinose tóxica secundária) que também representam diagnóstico diferencial da RPU. Apesar da inclusão nos critérios já descritos não há como garantir o diagnóstico de RPU. Isso porque talvez existam causas ainda não descritas ou não detectáveis de pseudorretinose pigmentar. Qualquer injúria celular poderia causar uma migração intrarretiniana de pigmento semelhante ao que ocorre nas degenerações retinianas hereditárias(5). O paciente também necessita ser acompanhado para excluir uma forma assimétrica da RPB.
A RPU é um diagnóstico raro e que ainda não possui consenso sobre seus fatores causadores, sua relação com a doença bilateral, sua característica hereditária e até mesmo sua existência. Acredita-se, atualmente, que muitos casos publicados como RPU seriam na verdade pseudorretinoses. Desta forma o médico oftalmologista deve estar atento para este diagnóstico diferencial.
REFERÊNCIAS
1. Mehra KS. Unilateral retinitis pigmentosa. Br J Ophthalmol. 1962;46(5):310.
2. Joseph R. Unilateral retinitis pigmentosa. Br J Ophthalmol. 1951;35:98-113.
3. Dreisler KK. Unilateral retinitis pigmentosa; two cases. Acta Ophthalmol. 1948;26(3): 385-93.
4. Farrell DF. Unilateral retinitis pigmentosa and cone-rod dystrophy. Clin Ophthalmol. 2009;3:263-70.
5. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-809.
6. Spadea L, Magni R, Rinaldi G, Dragani T, Bianco G. Unilateral retinitis pigmentosa: clinical and electrophysiological report of four cases. Ophthalmologica. 1998;212(5):350-4.
7. François J, Verriest G. Rétinopathie pigmentaire unilateral. Ophthalmologica. 1952; 124(2):65-88
8. Kato K, Miyake Y, Matsubara H, Uji Y. . Nihon Ganka Gakkai Zasshi, 2012;116(11):1086-93. Japanese.
9. Lommatzsch PK, Jensen OA, Prause JU, Bauke G. . Klin Monbl Augenheilkd. 1988;193(1):69-74. German.
10. Dhermy P, Raynaud G, Coscas G. . Arch Ophtalmol Rev Gen Ophtalmol. 1971;31(12):845-58. French.
11. Sekimoto M, Hayasaka S, Noda S, Setogawa T. Pseudoretinitis pigmentosa in patients with systemic lupus erythematosus. Ann Ophthalmol. 1993;25(7):264.
 Endereço para correspondência:
Endereço para correspondência:
Daniela Fernandes de Carvalho Rios
Av. Francisco Deslandes, 780/1.803 Belo Horizonte (MG)
30310-530 - Brazil
E-mail: [email protected]
Aceito para publicação: 17 de outubro de 2013
Submetido para publicação: 13 de março de 2013
Financiamento: Não houve financiamento para este trabalho.
Divulgação de potenciais conflitos de interesse: D.F.C.Rios, Nenhum; L.F.S.A.Carneiro, Nenhum; A.A.F.Cunha, Nenhum; C.B.M.Dalle, Nenhum; M.Frasson, Nenhum.
Estudo realizado na Clínica de Olhos da Santa Casa de Belo Horizonte (MG), Brasil.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.