Arq. Bras. Oftalmol. 2001; 64 (5): 10.1590/S0004-27492001000500016
Total: 2915
Maria Cristina Silvestrim1; Déborah Ferreira2; Jacob Cohen3
DOI: 10.1590/S0004-27492001000500016
RESUMO
Os autores relatam um caso de paciente do sexo masculino, 27 anos, com síndrome de Alport. Anteriormente ao tratamento cirúrgico do déficit visual por facoemulsificação e implante de lente intra-ocular, o paciente tinha sido submetido a transplante renal com recuperação total da função do órgão. Acuidade visual do paciente após o tratamento foi de 20/20 em ambos os olhos. Descrevem sucintamente a etiopatogenia, sinais clínicos e critérios diagnósticos da síndrome.
Descritores: Nefrite hereditária; Nefrite hereditária; Nefrite hereditária; Acuidade visual; Relato de caso
ABSTRACT
The authors report the case of a 27-year-old male patient with Alport syndrome. Prior to the surgical treatment of the visual deficit by phacoemulsification of the lens and IOL implantation, the patient had been submitted to renal transplantation with total recovery of renal function. Visual acuity post-surgery was 20/20 in both eyes. The authors present a short description of the etiopathology, clinical signs and diagnostic criteria of the syndrome.
Keywords: Hereditary nephritis; Hereditary nephritis; Hereditary nephritis; Visual acuity; Case report
INTRODUÇÃO
Nefrite hereditária ou Síndrome de Alport é a forma progressiva de doença glomerular, comumente associada à alterações do cristalino e perda da audição(1-2). É definida como glomerulonefrite não-imune hematúrica hereditária, caracterizada ultraestruturalmente pela espessura irregular, adelgaçamento e delaminação da membrana basal (MB) glomerular e tubular. É mais freqüente em homens(3). Os sinais clínicos assim como os critérios diagnósticos, são variáveis, tornando difícil definir exatamente o que é a síndrome(4).
O diagnóstico da síndrome é suspeitado em familiares com história de insuficiência renal, surdez e perda visual. No entanto, em 15% dos casos não há história familiar e o diagnóstico é feito através de biópsia renal(5).
A Síndrome de Alport classifica-se em seis tipos, no entanto as alterações oculares são restritas aos tipos juvenil I, II e VI, mas podem não estar presentes em todos os casos(4).
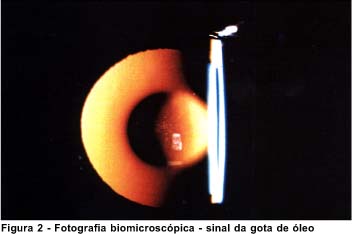
Alterações oculares - O lentecone é a alteração ocular patognomônica da síndrome. Corresponde a uma protusão da estrutura do cristalino em forma cônica (Figuras 1 e 2). Está associado à catarata subcapsular posterior em 80% dos casos(6). Várias teorias tem sido propostas para explicar o desenvolvimento do lentecone. Bach e Hess, 1898, sugeriram que a cápsula posterior fina por erro no desenvolvimento embrionário, resultasse em lentecone posterior. Von Hippel sugeriu que a inflamação vítrea fosse a causa primária(6). Atualmente, a fragilidade da cápsula tem sido apontada como causa do lentecone progressivo e catarata subcapsular posterior. O defeito pode estar presente nas MB de todo o corpo. Esta evidência sugere uma doença hereditária comum das MB(7). Além do lentecone, podem ser associadas à síndrome, a catarata subcapsular, rotura da capsula, esferofacia, miopia, atrofia da íris, síndrome da dispersão pigmentar, anisocoria, arco senil e alterações retinianas(7).
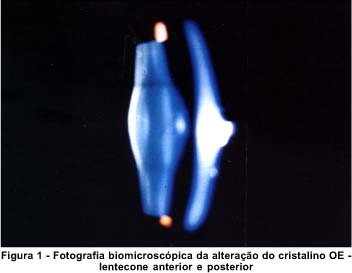
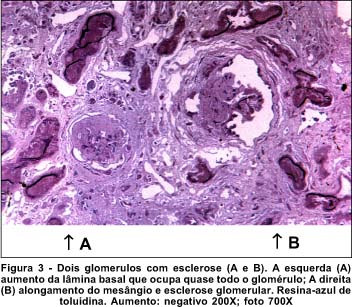
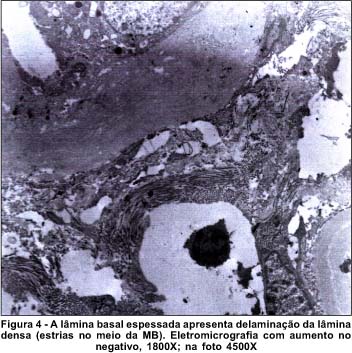
Relato do caso - EGS, 27 anos, sexo masculino, natural da cidade de Tefé - AM. Em 1985 iniciou com quadro clínico de edema progressivo de membros inferiores, anemia, polidipsia, oligúria, hipoacusia e baixa da acuidade visual (AV) principalmente no olho esquerdo. Não houve melhora do quadro inicial com tratamento clínico. Evoluiu com Síndrome urêmica e diminuição acentuada da AV. A biópsia renal (rim direito) revelou à microscopia óptica, esclerose glomerular difusa, alongamento do mesângio, atrofia tubular de alguns glomérulos e proliferação da membrana basal (Figura 3). As eletromicrografias demonstraram grande espessura da MB e fibrose intersticial do colágeno, compatíveis com Síndrome de Alport (Figura 4).
A história familiar registrou que 3 tios maternos apresentaram insuficiência renal associada às alterações auditivas e visuais.
O exame oftalmológico revelou: AV OD 20/100; OE 20/200. Biomicroscopia OD catarata capsular anterior e lentecone posterior moderado; OE lentecone anterior e posterior (Figuras 1 e 2). Oftalmoscopia OD alterações vasculares atróficas na região peripapilar; OE observação prejudicada, por aberrações astigmáticas dos meios transparentes. Tonometria OD 12 mmHg e OE 14 mmHg.
Ecografia: câmara anterior OD = 3,79 mm; OE = 2,59 mm; cristalino OD = 3,29 mm; OE = 4,39 mm; segmento posterior OD = 16,97mm; OE = 17,23mm.
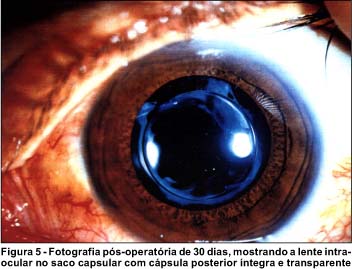
Tratamento: iniciou hemodiálise após o quadro de uremia, permanecendo em tratamento até junho de 1995, quando submeteu-se a transplante do rim direito (doador vivo - irmã) com total recuperação da função renal, mas persistindo a baixa da acuidade visual. Em 1997 o paciente submeteu-se a tratamento cirúrgico - facoemulsificação com implantação de lente intraocular de 5 mm de diâmetro, no saco capsular (Figura 5), inicialmente OE e após 30 dias o OD, resultando em AV 20/20 em ambos os olhos.
DISCUSSÃO
As alterações oculares na Síndrome da Alport, parecem estar limitadas às famílias com nefrite do tipo juvenil, com perda auditiva(4). Em contraste com a perda auditiva, comum na Síndrome, o lentecone anterior é menos freqüente, mas é patognomônico.
Presume-se que a patogênese da doença ocorra por defeito do tipo IV da MB do glomérulo(8-9). Na realidade o defeito primário está associado a herança ligada ao cromossomo X, em 85% dos casos(10) como resultado de mutações do gene COL 4A5(10-11). Estima-se que 2% a 5% dos casos do sexo masculino e 1% do sexo feminino, com insuficiência renal crônica (IRC) seja devido a Síndrome de Alport cuja prevalência varia de 1:5000 a 1:10000 nos EUA, no entanto estimativas mundiais ainda não são possíveis de serem determinadas(4). A doença não está associada a nenhum grupo racial ou regiões geográficas(5) . As manifestações iniciais da nefrite hereditária podem ser a hematúria assintomática - sinal cardinal da Síndrome - e/ou proteinúria. Inicia-se geralmente na infância, mas pode ser diagnosticada no adulto. A falência renal ocorre freqüentemente (85%)(12) em pacientes do sexo masculino entre 16 e 35 anos. Pessoas do sexo feminino são na sua maior parte, portadores sãos(3-5). A perda auditiva, ocorre comumente na infância, iniciando com a perda dos sons agudos, podendo progredir para a surdez total em 30% a 50% dos casos(13).
A evolução das manifestações oculares, parece guardar relação com a deterioração da função renal(14).
A imagem biomicroscópica do lentecone (sinal da gota de óleo) mostrado na figura 2 deste caso e registrado na ecografia mostra a medida antero-posterior do cristalino (espessura),do olho esquerdo significativamente maior, o que produz a imagem do sinal referido (Figura 2).
O prognóstico do paciente com Síndrome de Alport é pior em jovens do sexo masculino. A história familiar do paciente revelou que três tios maternos apresentaram insuficiência renal associada às alterações auditivas e visuais mas que faleceram sem o diagnóstico e tratamento adequados. Apesar da Síndrome ser rara (1:10000), a publicação deste caso se justifica pelo fato de o paciente ter história clínica detalhada, farta documentação da evolução das alterações, propedêutica completa com resultado de biópsias a nível de microscopia óptica e eletromicrografias e fundamentalmente ter se submetido a transplante renal com sucesso, garantindo-lhe uma sobrevida bastante grande, para poder usufruir de técnica cirúrgica capaz de recuperar totalmente a sua função visual.
REFERÊNCIAS
1. Grunfeld JP. The clinical spectrum of hereditary nephritis. Kidney Int 1985; 27:83-92.
2. Kashtan CE, Michael AF. Alport syndrome. Kidney Int 1996;50:1445-63.
3. Burton DR. Hereditary nephritis (Alport's syndrome): update.
4. Hereditary Nephritis Foundation (HNF). HNF annual meeting. HNF Newsletter (serial online) set.1995. Avaliable from URL: http://www.cc.utah.edu/~cla6202/HNF27.htm
5. National Institute of Deafness and other Communication Disorders (INDCD). Alport syndrome. Available from URL: http://www.nidcd.nih.gov/health/glossary/glos1.htm
6. The Boys Town Research Registry for Hereditary Hearing Loss. Alport Syndrome. Avaliable from URL: http://www.boystown. org/btnrh/deafgene.reg/alport_r.htm
7. Teekhasaenee C, Ninmanit S, Wutthiphaan S, Vareesangthiip K, Laohapand T, Malasitr P et al. Posterior polimorphous dystrophy and Alport syndrome. Ophthalmology 1991;98:1207-15.
8. Rumpelt HJ. Hereditary nephropathy (Alport's syndrome): correlation of clinical data with glomerular basement membrane alterations. Clin Nephrol 1980;13:203-7.
9. Gregory CM, Atkin LC. Alport's syndrome, Fabry's disease and a mail-patella syndrome. Hereditary Dis 1996;3:561-74.
10. Heiskari N, ZHANG X, Zhieu J, Leinonin A, Barker D, Gregory M et al. Identification of 17 mutations in the axons in the COL 4A5 collagen gene, but no mutations found un four axons in COL46: A study of 250 patients with hematuria and suspected of having Alport syndrome. J Am Soc Nephrol 1996;7:702-9.
11. Antignae C, Knebelmann B, Drouot L, Gros F, Deschenos G, Horscaula MC et al. Deletions in the COL 4A5 collagen gene in X-linked Alport syndrome. Characterization of the pathological transcripts in non renal cells: correlation with disease expression. J Clin Invest 1994;93:1195-207.
12. Trindade FLC, Katina J. Lentecone anterior na síndrome de Alport: reabilitação visual após facectomia extracapsular em cristalino transparente com implantação de lente intra-ocular de câmara posterior . Arq Bras Oftalmol 1989;52:132 .
13. Goldchmit M, Almeida GV de. Correlação entre o comprometimento renal e as alterações auditivas e oculares em pacientes portadores de síndrome de Alport. . Arq. Bras Oftamol 1989;52:140. .
14. Siqueira WCA de, Rosatelli Neto JM. Achados oftalmológicos na síndrome de Alport: A importância do oftalmologista no diagnóstico. . Arq Bras Oftalmol 1989;52:151-2. .
1 Médica Oftalmologista do Instituto de Oftalmologia de Manaus.
2 Médica Residente de Nefrologia - Faculdade de Ciências da Saúde - Universidade do Amazonas.
3 Professor Adjunto Doutor do Departamento de Clínica Cirúrgica - Universidade do Amazonas.
Endereço para correspondência: Dr. Jacob Cohen - Av. 7 de Setembro, 1613 - Manaus (AM) CEP 69005-141.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.