Arq. Bras. Oftalmol. 2012; 75 (4): 10.1590/S0004-27492012000400013
Total: 1541
Luciano Mesquita Simão1
DOI: 10.1590/S0004-27492012000400013
ABSTRACT
Neuromyelitis optica antibody (or aquaporin-4 antibody) is a well stablished serum marker associated to high-risk neuromyelitis optica syndrome that presents as an inflammatory demyelinating disease characterized by the occurrence of bilateral and simultaneous optic neuritis without complete visual recovery or it occurs as an isolated episode of transverse myelitis accompanied by longitudinally extensive spinal cord lesions. On the other hand, Leber hereditary optic neuropathy is a primarily hereditary disorder that affects all tissues of the body and its clinical presentation is tissue-specific for the optic nerve and, eventually, it might reach the spinal cord. Overlapping clinical features of neuromyelitis optica and Leber hereditary optic neuropathy may suggest common target organ diseases. The case report described herein emphasizes the coexistence of serum markers of both diseases, and suggests that further investigation of this challenging clinical presentation is warranted to confirm or rule out this association.
Keywords: Neuromyelitis optic; Optic atrophy, hereditary, Leber; Optic neuritis; DNA, mitochondrial; Mutation; Humans; Male; Adult; Case reports
RESUMO
Anticorpo da neuromielite óptica (ou anticorpo aquaporina-4) é um marcador sorológico bem estabelecido associado à síndrome de alto risco para neuromielite óptica, doença inflamatória desmielinizante, caracterizada por ocorrência bilateral, simultânea de neurite óptica ou por episódio isolado de mielite transversa com achado de lesões espinais longitudinais extensas. Por outro lado, a neuropatia óptica hereditária de Leber é uma doença primariamente hereditária que afeta todos os tecidos do corpo e sua apresentação clínica envolve o nervo óptico e, eventualmente, a medula espinal. Aspectos clínicos comuns sugerem que neuromielite óptica e neuropatia óptica hereditária de Leber possam atingir os mesmos órgãos. O caso descrito enfatiza a coexistência de marcadores sorológicos das duas doenças e sugere a necessidade de investigação futura desta apresentação clínica atípica para confirmar ou não esta associação.
Descritores: Neuromielite óptica; Atrofia óptica hereditária de Leber; Neurite óptica; DNA mitocondrial; Mutação; Humanos; Masculino; Adulto; Relatos de casos
CASE REPORT
Neuromyelitis optica antibody in Leber hereditary optic neuropathy: case report
Anticorpo da neuromielite óptica em neuropatia óptica hereditária de Leber: relato de caso
Luciano Mesquita Simão
Physician, Neuro-Ophthalmologist Universidade Federal de Minas Gerais - UFMG - Belo Horizonte (MG), Brazil
ABSTRACT
Neuromyelitis optica antibody (or aquaporin-4 antibody) is a well stablished serum marker associated to high-risk neuromyelitis optica syndrome that presents as an inflammatory demyelinating disease characterized by the occurrence of bilateral and simultaneous optic neuritis without complete visual recovery or it occurs as an isolated episode of transverse myelitis accompanied by longitudinally extensive spinal cord lesions. On the other hand, Leber hereditary optic neuropathy is a primarily hereditary disorder that affects all tissues of the body and its clinical presentation is tissue-specific for the optic nerve and, eventually, it might reach the spinal cord. Overlapping clinical features of neuromyelitis optica and Leber hereditary optic neuropathy may suggest common target organ diseases. The case report described herein emphasizes the coexistence of serum markers of both diseases, and suggests that further investigation of this challenging clinical presentation is warranted to confirm or rule out this association.
Keywords: Neuromyelitis optic; Optic atrophy, hereditary, Leber; Optic neuritis; DNA, mitochondrial; Mutation; Humans; Male; Adult; Case reports
RESUMO
Anticorpo da neuromielite óptica (ou anticorpo aquaporina-4) é um marcador sorológico bem estabelecido associado à síndrome de alto risco para neuromielite óptica, doença inflamatória desmielinizante, caracterizada por ocorrência bilateral, simultânea de neurite óptica ou por episódio isolado de mielite transversa com achado de lesões espinais longitudinais extensas. Por outro lado, a neuropatia óptica hereditária de Leber é uma doença primariamente hereditária que afeta todos os tecidos do corpo e sua apresentação clínica envolve o nervo óptico e, eventualmente, a medula espinal. Aspectos clínicos comuns sugerem que neuromielite óptica e neuropatia óptica hereditária de Leber possam atingir os mesmos órgãos. O caso descrito enfatiza a coexistência de marcadores sorológicos das duas doenças e sugere a necessidade de investigação futura desta apresentação clínica atípica para confirmar ou não esta associação.
Descritores: Neuromielite óptica; Atrofia óptica hereditária de Leber; Neurite óptica; DNA mitocondrial; Mutação; Humanos; Masculino; Adulto; Relatos de casos
INTRODUCTION
High-risk neuromyelitis optica (NMO) syndrome is an inflammatory demyelinating disease characterized by the occurrence of bilateral and simultaneous optic neuritis without complete visual recovery or it occurs as an isolated episode of transverse myelitis accompanied by longitudinally extensive spinal cord lesions. On the other hand, Leber hereditary optic neuropathy (LHON) is a primarily hereditary disorder due to mutations of mitochondrial DNA (mtDNA) that affects all tissues of the body and its clinical presentation is tissue-specific for the optic nerve and, eventually, the spinal cord. Overlapping clinical features may suggest that NMO and LHON are both organ-specific diseases. Interestingly, the association of multiple sclerosis (MS) and LHON has been documented, but few reports have evidenced the coexistence of NMO and LHON(1,2).
To our knowledge, this is the first case of presumed high-risk syndrome for NMO, suggested by seropositivity of aquaporin-4 (AQP-4) antibody, occurring simultaneously with LHON. Only two classical NMO cases have been described before and they both describe patients with the complete spectrum of the disease (i.e. symptoms related to neuromyelitis and to optic neuritis), harboring LHON mtDNA mutations (1,2).
CASE REPORT
A 43-year-old man presented complaints of painless, subacute onset of blurry vision in both eyes for two months. The right eye was first affected and the fellow eye became involved in the following week. He noted his vision was progressively worsening over time. He denied any other visual symptoms. Past ocular history was unremarkable. Past medical history included diabetes and hypertension. He had two maternal uncles with low vision.
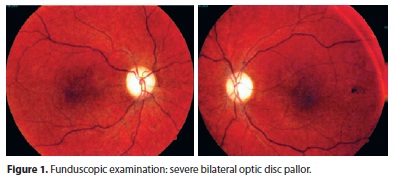
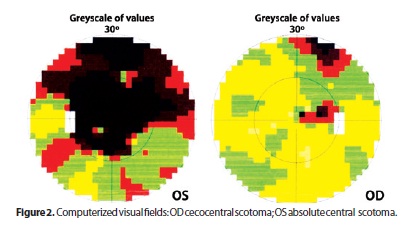
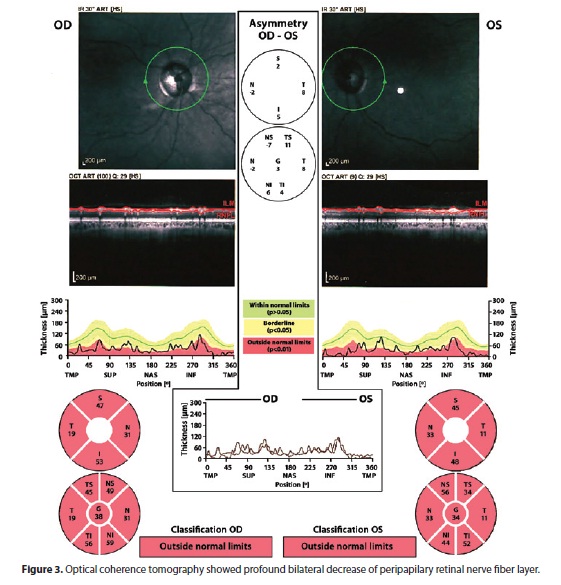
He underwent a first neuro-ophthalmic consultation two months after onset of symptoms. The visual acuity was 20/200 OD and counting fingers at 3 feet OS. He identified 8/11 Ishihara color plates OD and 0/11 OS. There was a left relative afferent pupillary defect. Slit lamp examination was unremarkable. Fundoscopic examination revealed bilateral disc pallor (Figure 1). Neurologic examination was normal. Computerized visual fields revealed cecocentral scotoma on the right and absolute central scotoma on the left (Figure 2). His family history of low vision lead us to conduct a blood test, which confirmed the existence of heteroplasmic primary 11778 LHON mtDNA mutation. Other family members denied undergoing neuro-ophthalmic examination or any blood tests. Serum analyses including ANA, rheumatoid factor, VDRL, viral serologies, B12, and folate were all negative or unremarkable. However, there was a positive title for AQP-4 antibody (1:80) and a cerebrospinal fluid examination showed high total protein content (63 mg/dl) with no oligoclonal bands or pleocytosis. Brain and spinal cord MRI were normal. Not surprisingly, optical coherence tomography confirmed a profound bilateral decrease of the peripapilary retinal nerve fiber layer (Figure 3). He underwent a five-day course of intravenous methylprednisolone with no visual improvement.
DISCUSSION
Neuromyelitis optica has been distinguished from MS by the presence of optic neuritis that is usually bilateral, simultaneous, without complete visual recovery often followed by permanent visual loss. Clinical findings that are also suggestive of NMO are longitudinally extensive spinal cord lesions with absent or unspecific brain imaging abnormalities, and often rapid progression to debility, and even death(3,4).
High-risk syndrome for NMO should be suspected in isolated recurrent bilateral optic neuritis, or in recurring isolated longitudinally-extensive myelitis(5). Those patients are in high risk of developing the second index event that characterizes the full-spectrum of the disease. This syndrome is also recognized as forme fruste or limited form of NMO. To help confirm the diagnosis there is also a serum antibody marker available named aquaporin-4 antibody. It binds selectively to the AQP4 water channel, a protein complex located in the astrocytic foot processes at the blood brain barrier. Because of the location of the antigen, it might be considered not only a marker, but actually the causative agent(4). It should be noted that anti-AQP4 is the first water channel specific autoantibody to be identified. AQP4 is considered the predominant water channel in the brain and spinal cord, and also in the skeletal muscles(4,5). The localization of AQP4 in the spinal cord is consistent with the immunopathology of NMO, which involves inflammation at both the gray and white matter regions, and for unknown reasons, it is largely restricted to the spinal cord and optic nerves(4).
It has also been well documented that the human antigen assay of anti-AQP4 provides a sensitivity of 85%, and a specificity of 100% in cases of isolated transverse myelitis or in cases of isolated optic neuritis, which are suggestive of high-risk syndromes for NMO(5).
In Brazil, the seroprevalence of anti-AQP4 test in NMO or high-risk syndrome for NMO patients was first reported in 2008 and the seropositivity in this group was 64.3%. This finding was similar to that observed in other countries(6).
Conversely, LHON is an mtDNA mutation disease whose clinical setting is confined to the optic nerve and, eventually, the spinal cord(7). Pathogenic mtDNA mutations occur mainly in the homoplasmic state (i.e. there is only mutant mtDNA within an individual). However, heteroplasmy is a mixture of a wild type and mutant mtDNA observed in about 15% of affected families(8, 9).
It is worth remembering that the severity of visual loss and the optic atrophy are unpredictable despite the mutation load. This can be attributed to the impossibility of biopsying the target tissues (i.e. the optic nerve and retina) since the quantification of heteroplasmy is currently available only from blood samples. Previous studies suggest the limited value of blood tests in predicting the degree of heteroplasmy in other tissues(9).
Although it has been suggested that the occurrence of 70% of mutant mtDNA in the blood of heteroplasmic individuals may contribute to optic atrophy, some reports have shown affected individuals with much lower mutation loads(10,11). It can be supposed that such patients should present enough mutant mtDNA load in the optic nerves to relapse the disease(11).
Seemingly NMO and LHON may share some clinical similarities but few reports have questioned the linkage between these diseases. Due to the paucity of LHON mtDNA mutation encountered in NMO patients, some studies have failed showing genetic correlation(7). However, this is the first case report to demonstrate NMO antibody in LHON, and probably, more similar cases will be reported as NMO antibody test has increasingly been ordered in medical practice.
In conclusion, the case described herein as painless, subacute bilateral decreased vision, and poor visual recovery, accompanied by maternal history of low vision and seropositivity of mtDNA mutation confirmed the diagnosis of LHON. Simultaneously the seropositivity of AQP-4 antibody suggests co-existence of a marker that labels individuals with NMO or high-risk syndrome for NMO. Whether this is a fact or mere coincidence only prospective studies would help confirm, or rule out, this striking correlation. Ophthalmologists and neurologists should bear in mind the possibility of this uncommon clinical association.
REFERENCES
1. Celebisoy N, Akyurekli O, Copur A. Devic's neuromyelitis optica: a case with mitochondrial DNA mutations. Eur Neurol. 2006;55(2):93-5.
2. Ghezzi A, Baldini S, Zaffaroni M, Leoni G, Koudriavtseva T, Casini AR, Zeviani M. Devic's neuromyelitis optica and mitochondrial DNA mutation: a case report. Neurol Sci. 2004;25(Suppl 4):S380-2.
3. Weinshenker BG. Neuromyelitis optica: what it is and what it might be. Lancet. 2003; 361(9361):889-90. Erratum in: Lancet. 2003;362(9383):582.
4. Cross SA. Rethinking neuromyelitis optica (Devic disease). J Neuroophthalmol. 2007; 27(1):57-60.
5. Lana-Peixoto MA. Devic's neuromyelitis optica: a critical review. Arq Neuropsiquiatr. 2008;66(1):120-38.
6. Adoni T, Lino AM, Marchiori PE, Kok F, Callegaro D. Seroprevalence of NMO-IgG antibody in Brazilian patients with neuromyelitis optica. Arq Neuropsiquiatr. 2008; 66(2B):295-7.
7. Hudson G, Mowbray C, Elson JL, Jacob A, Boggild M, Torroni A, et al. Does mitochondrial DNA predispose to neuromyelitis optica (Devic's disease)? Brain. 2008;131(Pt 4): e93.
8. Puomila A, Viitanen T, Savontaus ML, Nikoskelainen E, Huoponen K. Segregation of the ND4/11778 and the ND1/3460 mutations in four heteroplasmic LHON families. J Neurol Sci. 2002;205(1):41-5.
9. Huoponen K, Puomila A, Savontaus ML, Mustonen E, Kronqvist E, Nikoskelainen E. Genetic counseling in Leber hereditary optic neuropathy (LHON). Acta Ophthalmol Scand. 2002;80(1):38-43.
10. Howell N, Xu M, Halvorson S, Bodis-Wallner I, Sherman J. A heteroplasmic LHON family: tissue distribution and transmission of the 11778 mutation. Am J Hum Genet. 1994;55(1):203-6.
11. Jacobi FK, Leo-Kottler B, Mittelviefhaus K, Zrenner E, Meyer J, Pusch CM, et al. Segregation patterns and heteroplasmy prevalence in Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2001;42(6):1208-14.
 Correspondence address:
Correspondence address:
Luciano M. Simão.
Av. do Contorno, 4747 - Conj. 1705
Belo Horizonte - MG - 30110-090 - Brazil
E-mail: [email protected]
Submitted for publication: June 20, 2011
Accepted for publication: March 21, 2012
Study carried out at Institution: Visio Clínica Oftalmológica e Oftalmoclínica Rui Marinho.
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: L.M.Simão is employee of Universidade Federal de Minas Gerais.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.