Arq. Bras. Oftalmol. 2011; 74 (6): 10.1590/S0004-27492011000600014
Total: 2377
Fabio Yamasato Yonamine1; Tiago Eugênio Faria e Arantes1; Cristina Muccioli1
DOI: 10.1590/S0004-27492011000600014
ABSTRACT
Fabry disease is an X-linked disease of glycosphingolipid (GL) metabolism. The accumulation of GL in tissues can affect multiple organ systems. Initial symptoms includes episodes of severe pain in the extremities, cornea verticillata and skin lesions. In late stages, kidney, heart and brain can be involved. This report attempts for importance of cornea verticillata in the diagnosis of Fabry disease in a young patient with pain in the lower limb extremities.
Keywords: Fabry disease; Corneal opacity; Case report
RESUMO
A doença de Fabry é um erro inato no metabolismo de glicosfingolipides (GL) ligado ao cromossomo X. O acúmulo de GL nos tecidos pode afetar múltiplos órgãos e sistemas. Os sintomas iniciais incluem episódios de dor nas extremidades, córnea verticilata e lesões na pele. Em estágios avançados, os rins, coração e cérebro podem ser envolvidos. Este relato de caso enfatiza para importância da córnea verticilata para o diagnóstico de doença de Fabry em paciente jovem com dor nos membros inferiores.
Descritores: Doença de Fabry; Opacidade da córnea; Relato de caso
CASE REPORTS RELATOS DE CASOS
Fabry disease - importance of screening in cornea verticillata: case report
Doença de Fabry - importância do rastreamento em córnea verticilata: relato de caso
Fabio Yamasato Yonamine; Tiago Eugênio Faria e Arantes; Cristina Muccioli
Physician, Ophthalmology Department, Universidade Federal de São Paulo - UNIFESP - São Paulo (SP), Brazil
ABSTRACT
Fabry disease is an X-linked disease of glycosphingolipid (GL) metabolism. The accumulation of GL in tissues can affect multiple organ systems. Initial symptoms includes episodes of severe pain in the extremities, cornea verticillata and skin lesions. In late stages, kidney, heart and brain can be involved. This report attempts for importance of cornea verticillata in the diagnosis of Fabry disease in a young patient with pain in the lower limb extremities.
Keywords: Fabry disease/diagnosis; Corneal opacity; Case report
RESUMO
A doença de Fabry é um erro inato no metabolismo de glicosfingolipides (GL) ligado ao cromossomo X. O acúmulo de GL nos tecidos pode afetar múltiplos órgãos e sistemas. Os sintomas iniciais incluem episódios de dor nas extremidades, córnea verticilata e lesões na pele. Em estágios avançados, os rins, coração e cérebro podem ser envolvidos. Este relato de caso enfatiza para importância da córnea verticilata para o diagnóstico de doença de Fabry em paciente jovem com dor nos membros inferiores.
Descritores: Doença de Fabry/diagnóstico; Opacidade da córnea; Relato de caso
INTRODUCTION
Fabry disease (FD) is an X-linked disease caused by deficient activity of lysosomal enzyme α-galactosidase A (α-Gal A). Glycosphingolipids (GL) accumulate in the lysosomes of various cells (eg, in the vascular endothelium of multiple organs) owing to α-Gal A deficiency. The accumulation of GL in the lysosomes causes cellular dysfunction; this, in turn, triggers cells cascade and tissue ischemia and fibrosis. The incidence of Fabry disease is estimated to be approximately 1:50,000 males(1).
The first clinical manifestations of the disease, which consist of episodes of severe pain in the extremities (acroparesthesias), corneal changes and skin lesions (angiokeratoma) develop in the childhood or early adolescence. In the third and fourth decades, the progressive deposition of GL in blood vessels and smooth muscles leads to cardiovascular changes like angina pectoris and congestive heart failure. Other features like a renal failure, stroke and hypoacusis may also occur(2,3). Elderly heterozygotes female carriers occasionally develop related symptoms similar to male patients(4).
Cornea verticillata is the most frequent ocular finding of Fabry disease. Women are carriers and, in most cases, a vortex keratopathy pattern is found. Visual impairment is an uncommon finding. Lens opacities may occur with granular deposits in posterior capsule. Aneurysmal dilatation and tortuosity of conjunctival and retinal vessels may also occur(5,6).
Treatment is composed of medications to alleviate symptoms as also disease-specific therapy to delay and prevent possible serious organ damage. Enzyme reposition therapy (ERT) provides the biologically functional protein to the patient. The goals of therapy are improvement of symptoms and prevention of disease complications. Multiple clinical trials with recombinant a-Gal A have been performed to investigate the safety and efficacy of ERT in patients with Fabry disease. The rate of progression of renal, cardiac, and cerebrovascular complications and death among patients who received active drug was reduced compared with the placebo group. Current recommendations suggest that ERT should be initiated as early as possible in all males with Fabry disease(4,6).
CASE REPORT
A 16 year-old boy was referred to the Department of Ophthalmology of the Universidade Federal de São Paulo - UNIFESP during investigation of Marfan Syndrome. He was under evaluation complaining of pain and paresthesia in both feet for six months. He had been previously evaluated by a pediatrician and performed a rheumatologic screening with negative results. He had no ophthalmological complaints and his past medical and ocular history were unremarkable. He also had no familial history of genetic disorders or ocular diseases.
The ophthalmological exam revealed uncorrected visual acuity of 20/20 in both eyes and slit-lamp evaluation disclosed bilateral cornea verticillata (Figure 1). Careful examination of anterior and posterior segments did not show any other finding with no signs to support the diagnosis of Marfan Syndrome based on the ophthalmological evaluation.
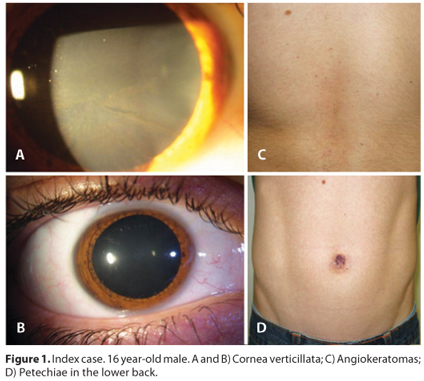
He never used any medication that could cause corneal deposits, such as amiodarone, chloroquine, indomethacin, and phenothiazines.
A comprehensive physical examination was performed revealing angiokeratomas around the navel and petechiae in his back and arms.
Fabry disease was suspected and the patient was referred for genetic evaluation. The low activity of α-Gal A associated with clinical manifestation confirmed the diagnosis.
The family was invited for ophthalmic evaluation. The father had a normal ocular exam, but the examination of the mother revealed vortex keratopathy (carrier) and his brother had cornea verticillata and angiokeratomas with no other signs or symptoms of the disease (Figure 2).
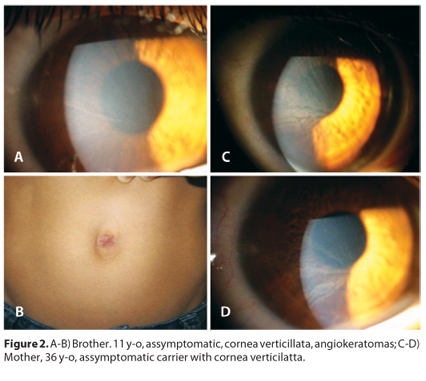
DISCUSSION
In this case, the disclosure of cornea verticillata was decisive for the diagnosis of Fabry disease, and led to the diagnosis of his brother and his mother. In the Fabry Outcome Survey registry, cornea verticillata was present in 77% of females and 73% of males undergoing detailed ophthalmic examination(3). Slit-lamp examination reveals cornea verticillata, which manifests as whirl-like white-to-golden-brown opacities that extend from the center to the periphery of the cornea. Vision is not usually impaired. Cornea verticillata is often prominent in female carriers and play an important role in the early recognition of Fabry disease.
Lenticular changes are present in approximately 30% of affected males and include a characteristic pattern of granular anterior capsular or subcapsular deposit (Fabry cataract). The cataracts, which are best observed through a dilated pupil slit-lamp examination using retroillumination are whitish, with spoke-like deposits of fine granular material on or near the posterior lens capsule. These lines usually radiate from the central part of the posterior cortex(3). Aneurysmal dilatation and tortuosity of conjunctival and retinal vessels may also occur. Data from Fabry Outcome Survey indicate that vessel tortuosity is observed more frequently in individuals with a higher disease severity score(3).
When the familial history is unknown, the diagnosis becomes difficult and frequently delayed, leading to retardation of enzyme replacement therapy and an increased morbidity due to cardiac, renal and brain complications. Some patients die from stroke, heart attack and congestive heart failure secondary to aneurismal changes caused by Fabry disease without diagnosis(7,8).
The α-Gal A activity may be measured in plasma, serum, leukocytes, tissue biopsies and cultured skin fibroblasts. In males, the most efficient and reliable method for the diagnosis of Fabry disease is the demonstration of deficient α-Gal A enzyme activity in plasma, isolated leukocytes, and/or cultured cells. In females, measurement of α-Gal A enzyme activity is unreliable. Many female carriers have normal α-Gal A enzyme activity, although, demonstration of decreased α-Gal A enzyme activity is diagnostic of the carrier state. The screening for female carriers can be done with the ophthalmic examination based on the presence of cornea verticillata present in 70-90% of females carrier(5,9).
CONCLUSION
Cornea verticillata is a relatively common ocular finding, typically presenting with no associated visual impairment. However, young patients should be investigated for possible genetic disorders especially when there is no history of use of medications related with corneal deposits. With early diagnosis of Fabry disease, enzyme replacement therapy can be instituted, avoiding serious complications and decreasing the morbidity and mortality associated with the disease.
REFERENCES
1. Mehta A, Ricci R, Widmer U, Linhart A, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34(3):236-42.
2. Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30(2):184-92
3. Sodi A, Ioannidis AS, Mehta A, Davey C, Beck M, Pitz S. Ocular manifestations of Fabry's disease: data from the Fabry Outcome Survey. Br J Ophthalmol. 2007;91(2):210-4.
4. Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338-46.
5. Gupta S, Ries M, Kotsopoulos S, Schiffmann R. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. Medicine (Baltimore). 2005;84(5):261-8.
6. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. International Collaborative Fabry Disease Study Group. Safety and efficacy of recombinant human alpha-galactosidase A - replacement therapy in Fabry's disease. N Engl J Med. 2001;345(1):9-16.
7. Nakao S, Kodama C, Takenaka T, Takenaka A, Yasumoto Y, Yoshida A, et al. Fabry disease: detection of underdiagnosed hemodialysis patients and identification of a "renal variant" phenotype. Kidney Int. 2003; 64(3):801-7.
8. Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, Mckenna WJ, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105(12):1407-11.
9. Cordeiro CA, Oréfice F, Lasmar EP, Santos HH, Valadares ER. Córnea verticilata - marcador clínico da doença de Fabry: relato de caso. Arq Bras Oftalmol. 2007;70(4):701-5.
 Correspondence address:
Correspondence address:
Fabio Yamasato Yonamine
Rua Botucatu, 822
São Paulo (SP) - 04023-062 - Brazil
E-mail: [email protected]
Submitted for publication: May 27, 2010
Accepted for publication: August 8, 2011
Study carried out at Departamento de Oftalmologia, Universidade Federal de São Paulo - UNIFESP - São Paulo (SP), Brazil.
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: F.Y.Yonamine, None; T.E.Faria e Arantes, None; C.Muccioli, None.
Editorial Note: After completing the confidential analysis of the manuscript, ABO discloses, with her agreement, the name Dr. Diane Ruschel Marinho as a reviewer. We thank her effort and expertise in participating in this process.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.