Arq. Bras. Oftalmol. 2007; 70 (4): 10.1590/S0004-27492007000400021
Total: 2425
Ellen Carrara Fonseca1; Lígia Issa De Fendi2; Paulo Sérgio Andretta3; Rosana Teresa Alves Lois Martin4; José Augusto Alves Ottaiano5
DOI: 10.1590/S0004-27492007000400021
RESUMO
Apresentação de um caso de síndrome de Urbach-Wiethe com manifestações típicas, inclusive oculares. Paciente do sexo feminino, 15 anos, com quadro de prurido ocular relacionado à presença de lesões papuliformes em margens palpebrais (blefarose moniliforme), em associação com outras alterações sistêmicas. O diagnóstico foi confirmado por meio de biópsia cutânea e foi instituído uso de lágrimas artificiais, com alívio parcial do sintoma. O objetivo do trabalho é relatar um caso com manifestações características da doença atendido no Setor de Oftalmologia da Faculdade de Medicina de Marília.
Descritores: Proteinose lipóide de Urbach e Wiethe; Lipidoses; Pálpebras; Síndrome; Manifestações oculares; Relatos de casos
ABSTRACT
We present a case of Urbach-Wiethe syndrome with typical findings, including ocular lesions. A 15-year-old girl was referred to our department complaining of itchy eyelid lesions (moniliform blepharosis) associated with other systemic manifestations. Diagnosis was confirmed by performing skin biopsy. Artificial tears were prescribed, with partial relief of the symptom. The objective of the present study is to describe a typical case of Urbach-Wiethe syndrome attended at the Ophthalmologic Sector of the Medical School of Marília.
Keywords: Lipoid proteinosis of Urbach and Wiethe; Lipidoses; Eyelids; Syndrome; Eye manifestations; Cases report
INTRODUÇÃO
A síndrome de Urbach-Wiethe, também conhecida como Lipoidoproteinose, Hialinose Cutâneo-Mucosa, Lipoglicoproteinose e Proteinose cutâneo-mucosa é doença autossômica recessiva rara caracterizada pela deposição de material hialino em laringe, membranas mucosas, pele, olhos, cérebro e outros órgãos, causando manifestações sistêmicas típicas(1-8). Manifestações oculares incluem depósitos hialinos em margem palpebral, conjuntiva, córnea, trabeculado e membrana de Bruch. As lesões palpebrais estão presentes na maioria dos casos e são patognomônicas, sendo importantes para um diagnóstico imediato(1,9-10).
A primeira descrição data de 1908, por Seibenmann, mas o primeiro relato detalhado foi feito em 1929 pelo dermatologista Erich Urbach e pelo otorrinolaringologista Camilo Wiethe(11).
Há, em todo o mundo, aproximadamente 300 casos relatados na literatura.
RELATO DE CASO
Paciente do sexo feminino, 15 anos, branca, estudante, natural de Porto Primavera-SP e procedência atual Marília-SP.
Vem encaminhada ao serviço de urgência e emergência do setor de oftalmologia da Faculdade de Medicina de Marília com queixa de prurido em ambos os olhos há alguns meses. Negava alteração de acuidade visual, dor, hiperemia, lacrimejamento ou secreção; sem antecedentes de patologia ocular prévia.
Segundo a mãe, a paciente apresenta, desde o nascimento, rouquidão, dispnéia noturna e respiração bucal. Por volta dos 9 meses de idade iniciou quadro de eritema em regiões subescapular e cervical posterior, evoluindo com lesões de pele ertitêmato-crostosas disseminadas, acompanhadas de prurido, com resolução do quadro em aproximadamente 5 anos, após tratamentos inespecíficos; permaneceu com cicatrizes e espessamento cutâneo em área de cotovelos e joelhos e em região cervical (Fotos 1 e 2). Relata desenvolvimento neuropsicomotor adequado; nega dificuldade de aprendizagem na escola.
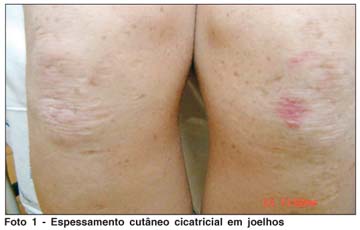
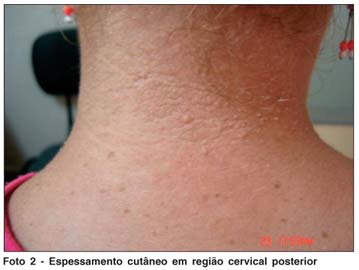
Mantém acompanhamento psiquiátrico há alguns meses, com diagnóstico de distimia. Faz uso de fluoxetina e bromazepam diários.
Não há história de consangüinidade conhecida na família. Pai, mãe, irmão e irmã são hígidos.
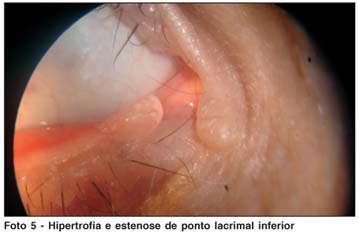
Ao exame oftalmológico apresentava acuidade visual sem correção de 20/20 em ambos os olhos. À biomicroscopia foram evidenciadas lesões papulosas ao longo das margens palpebrais superiores e inferiores, com estenose dos pontos lacrimais (Fotos 3, 4 e 5); não havia alterações em conjuntiva, córnea, íris e cristalino ou reação inflamatória em câmara anterior. A pressão intra-ocular era normal em ambos os olhos, bem como o exame de fundo de olho.
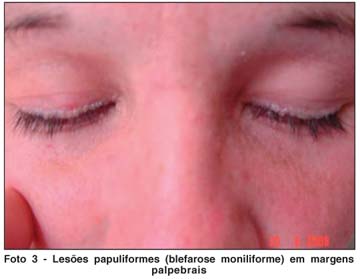
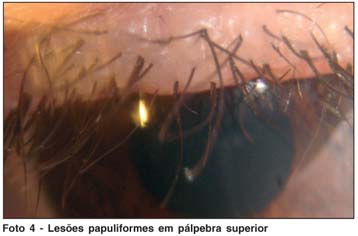
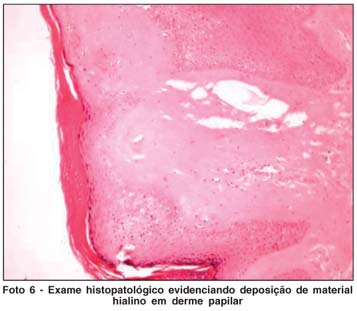
Biópsias de pele de regiões de tórax, axila e cotovelo confirmaram o diagnóstico de Síndrome de Urbach-Wiethe, evidenciando dermatose com papilomatose e deposição de material hialino na derme papilar (Fotos 6 e 7).
Ao exame de videofibronasofaringolaringoscopia apresentava espessamento epidérmico da epiglote, hiopertrofia de falsas cordas vocais, irregularidade da mucosa de cordas vocais, fenda fusiforme, constrição anteroposterior e língua endurecida.
Não houve alteração no eletroencefalograma.
Atualmente em uso de lágrimas artificiais, com alívio parcial dos sintomas.
DISCUSSÃO
A Doença de Urbach-Wiethe é uma rara desordem autossômica recessiva, de fisiopatologia ainda desconhecida, caracterizada por deposição de material hialino em vários órgãos. Este material é composto por ácido hialurônico e colesterol. Estudo realizado com vários pacientes na África do Sul identificou como responsável pela doença a mutação homozigótica Q276X no exon 7 do gene ECM1 (extracellular matrix protein 1(10,12)). O papel do gene ECM1 na pele de humanos não está esclarecido(13), entretanto, na epiderme, pode ter impacto na dinâmica de diferenciação dos queratinócitos, alterando seu padrão normal de maturação e levando à hiperqueratose. Na derme pode agir como "cola biológica" para glicosaminoglicanos e como fator de crescimento(14). A perda do ECM1 pode ter efeito na homeostase dermal causando infiltração cutânea e as alterações dermatopatológicas da hialinose.
Acomete igualmente homens e mulheres e é mais comum em europeus, principalmente holandeses e alemães(1), e em sul-africanos(10). Há alta incidência de consangüinidade(6,15). Ocorre tipicamente na infância, mas alguns casos manifestam-se na vida adulta.
Crianças costumam apresentar rouquidão precoce conseqüente ao depósito de material hialino na membrana mucosa das cordas vocais(5), evoluindo gradualmente com lesões cutâneas que cicatrizam espontaneamente em alguns anos; pode haver distúrbios de aprendizagem e alterações comportamentais decorrentes da presença de depósitos hialinos no sistema nervoso central.
As principais manifestações clínicas encontradas são:
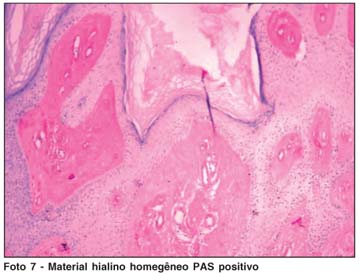
O diagnóstico é confirmado através de biópsia cutânea. O exame histopatológico evidencia espessamento da derme com extensos depósitos de material hialino homogêneo extracelular PAS positivos e diástase-resistentes, caracterizados por acantose, hiperqueratose e paraqueratose da pele(5,21). A reduplicação da lâmina basal perivascular sugere aumento da produção de colágenos tipos IV e V pelas células endoteliais vasculares e tipos I e II pelos fibroblastos.
Pacientes portadores da doença devem ser acompanhados por equipe multidisciplinar, não havendo tratamento específico. Procedimentos cirúrgicos como ressecção de cordas vocais e dermoabrasão em lesões cutâneas podem ser realizados a fim de amenizar sintomas. Há relato de um caso em que foi realizada dermoabrasão em região palpebral com laser de dióxido de carbono, com bons resultados a curto prazo(7). Não há relatos de seguimentos de pacientes a longo prazo, principalmente na área oftalmológica.
O curso da doença é geralmente crônico e benigno(8). A evolução é compatível com curso de vida normal, exceto quando complicada por obstrução de vias aéreas(6,8). A qualidade de vida dos pacientes pode ser afetada pelas lesões desfigurantes e pela rouquidão permanente(2).
O conhecimento das lesões típicas oculares pode ser de grande auxílio no diagnóstico precoce da doença, que, na maioria das vezes, é confirmado tardiamente.
REFERÊNCIAS
1. Blodi FC, Whinery RD, Hendricks CA. Lipoid-proteinosis (Urbach-Wiethe) involving the lids. Trans Am Ophthalmol Soc. 1960;58:155-66.
2. Sharma V, Kashyap S, Betharia SM, Gupta S, Pathak H. Lipoid proteinosis: a rare disorder with pathognomonic lid lesions. Clin Experiment Ophthalmol. 2004;32(1):101-2.
3. Rosenthal AR, Duke JR. Lipoid proteinosis: case report of direct lineal transmission. Am J Ophthalmol. 1967;64(6):1120-5.
4. Staut CC, Naidich TP. Urbach-Wiethe disease (lipoid proteinosis). Pediatr Neurosurg. 1998;28(4):212-4.
5. Lima LR, Mulinari-Brenner FA, Manfrinato LC, Dal Pizol AS, Serafini SZ, Fillus Neto J. Lipoid Proteinosis - a report of two cases. An Bras Dermatol. 2003;78(6):723-7.
6. Cordoro KM, Osleber MF, Shaffer JJ, De Leo V. Lipoid proteinosis. [last updated in July 21, 2006 [: about 10 p] [citado 2006 nov 12]. Available from: http://www.emedicine.com/DERM/topic241/htm
7. Rosenthal G, Lifshitz T, Monos T, Kachco L, Argov S. Carbon dioxide laser treatment for lipoid proteinosis (Urbach-Wiethe syndrome) involving the eyelids. Br J Ophthalmol. 1997;81(3):253.
8. Pinto JA, Faria CAR, Sallum RA, Marigo C. Manifestações otorrinolaringológicas na syndrome de Urbach-Wiethe (Lipoproteinose). Rev Bras Otorrinolaringol. 1988;54(2):57-9.
9. Sellami D, Masmoudi A, Turki H, Mseddi M, Kammoun B, Elleuch N, et al. [Ophthalmic manifestations of lipoid proteinosis]. Presse Med. 2006;35(5 Pt 1):796-8. French
10. Van Hougenhouck-Tulleken W, Chan I, Hamada T, Thornton H, Jenkins T, McLean WH, et al. Clinical and molecular characterization of lipoid proteinosis in Namaqualand, South Africa. Br J Dermatol. 2004;151(2):413-23.
11. Urbach E, Wiethe C. Lipoidosis cutis et mucosae. Virchows Archiv fur pathologische anatomie und physiologie und fur klinische medizin. 1929;273: 285-319.
12. Hamada T, Mclean WH, Ramsay M, Ashton GH, Nanda A, Jenkins T, et al. Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Hum Mol Genet. 2002;11(7):833-40.
13. Chan I. The role of extracellular matrix protein 1 in human skin. Clin Exp Dermatol. 2004;29(1):52-6.
14. Monglat M, Fu J, Oldershaw R, Greenhalgh R, Gown AM, Iozzo RV. Perlecan protein core interacts with extracellular matrix protein 1 (ECM1), glycoprotein involved in bone formation and angiogenesis. J Biol Chem. 2003;278(19):17491-9.
15. Costagliola C, Verolino M, Landolfo P, Winkler NR, Mastropasqua L, Landolfo V. Lipoid proteinosis (Urbach-Wiethe Disease). Ophthalmologica. 1999;213(6):392-6.
16. Zapata MA, Romera M, Linares F. Glaucoma juvenil associado a síndrome de Urbach-Wiethe. A propósito de um caso. Annais d'Oftalmol. 2004;12(2):92-4.
17. Irkec M, Orhan M, Orhan D, Durgun B, Can C. Dry eye Syndrome associated with Urbach-Wiethe disease. J Pediatr Ophthalmol Strabismus. 1996;33(5):265-8.
18. Johnson LN, Hepler RS. Corectopia and lipoid proteinosis. Br J Ophthalmol. 1989;73(5):394-6.
19. Maia R, Teixeira L, Grago J. Urbach-Wiethe disease/lipoidproteinosis. Acta Med Port. 1998;11(12):1113-5.
20. Kleinert R, Cervos-Navarro J, Kleinert J, Walter GF, Steiner H. Predominantly cerebral manifestation in Urbach-Wiethe's syndrome (lipoid proteinosis cutis et mucosae): a clinical and pathomorphological study. Clin Neuropathol. 1987;6(1):43-5.
21. Fabrizi G, Porfiri B, Borgioli M, Serri F. Urbach-Wiethe disease. Light and electron microscopic study. J Cutan Pathol 1980;7(1):8-20.
Endereço para correspondência:
Ellen Carrara Fonseca
Rua 24 de dezembro, 250
Marília (SP) - CEP 17500-090
E-mail: [email protected]
Recebido para publicação em 29.09.2006
Última versão recebida em 11.12.2006
Aprovação em 08.01.2007
Trabalho realizado no Departamento de Oftalmologia da Faculdade de Medicina de Marília - FAMEMA - Marília (SP) - Brasil.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.