
Heloisa Andrade Maestrini1; Luciene Chaves Fernandes2; Ana Cláudia Monteiro Oliveira3
DOI: 10.1590/S0004-27492004000600005
RESUMO
OBJETIVOS: Descrever o quadro clínico e os resultados dos exames complementares dos pacientes portadores das seguintes distrofias retinianas: amaurose congênita de Leber (ACL), acromatopsia, distrofia de cones e distrofia mista, atendidos no Serviço de Visão Subnormal do Hospital São Geraldo da UFMG, no período de 1992 a 2003. MÉTODOS: Análise retrospectiva dos prontuários de 40 pacientes, sendo 10 portadores de ACL, 17 com acromatopsia, 6 com distrofia de cones e 7 com distrofia mista. RESULTADOS: A acuidade visual foi extremamente baixa na ACL, variando de 20/710 a percepção luminosa. Situou-se em torno de 20/200 na acromatopsia, 20/280 na distrofia de cones e 20/260 na mista. A alta hipermetropia foi o erro refracional mais comum na ACL, ao passo que a hipermetropia predominou na acromatopsia e na distrofia de cones e a miopia na mista. A fundoscopia mostrou-se alterada na maioria dos casos de ACL, distrofia de cones e distrofia mista e normal na maioria dos acromatas. A compressão óculo-digital e o enoftalmo foram exclusivos da ACL, ao passo que a fotofobia e a dificuldade na discriminação de cores predominaram nos outros grupos. O nistagmo e o estrabismo foram freqüentes em todos eles. O atraso no desenvolvimento neuro-psico-motor foi muito freqüente no grupo da ACL e quase ausente nos demais. A ACL apareceu associada a síndromes genéticas em 2 casos. Os sintomas da ACL e da acromatopsia se manifestaram ao nascimento ou no 1º ano de vida, ao passo que na distrofia de cones e na mista surgiram também em idades mais avançadas, porém não depois dos 10 anos. A consangüinidade e a história familiar positiva foram altamente prevalentes em todos os grupos. O ERG mostrou ausência de resposta na ACL, redução da resposta fotópica na acromatopsia e na distrofia de cones e redução difusa na mista. Os testes de visão de cores mostraram alterações principalmente na acromatopsia e na distrofia de cones. CONCLUSÕES: As distrofias retinianas da infância são um grupo heterogêneo de doenças que se manifestam por meio de sintomas inespecíficos. Uma análise cuidadosa dos sintomas, o exame oftalmológico completo e os exames complementares, principalmente ERG, testes de visão de cores e campo visual, podem ser úteis em seu diagnóstico.
Descritores: Baixa visão; Fotorreceptores de vertebrados; Defeitos da visão cromática; Cones (retina); Retinite pigmentosa
ABSTRACT
PURPOSE: To describe the clinical features and the results of diagnostic methods in all patients with diagnosis of one of the following retinal dystrophies: Leber congenital amaurosis (LCA), achromatopsia, cone distrophy or cone-rod distrophy, examined at the Low Vision Department of the Federal University of Minas Gerais, in the period of 1992 to 2003. METHODS: Retrospective analysis of charts of 40 patients. Ten had LCA, 17 had achromatopsia, 6 had cone distrophy and 7 had cone-rod distrophy. RESULTS: Visual acuity was extremely low in patients with LCA, ranging from 20/710 to light perception. The mean value for achromatopsia was 20/200, 20/280 for cone distrophy and 20/260 for cone-rod distrophy. High hyperopia was the most common refractional error in LCA patients. Hyperopia was more frequent in cases of achromatopsia and cone distrophy, while in cone-rod distrophy myopia predominated. Fundoscopy was altered in most cases of LCA, cone distrophy and rod-cone distrophy, and normal in most cases of achromatopsia. Oculodigital sign and enophtalmus were found only in LCA patients while photofobia and color vision defects prevailed in other groups. Nistagmus and strabismus were frequent findings in all groups. There was a high incidence of delayed neuro-psycho-motor development in LCA patients. Two of them had also associated genetic syndromes. Patients presented symptoms very early in life in LCA and achromatopsia, while in cone and cone-rod distrophies symptoms appeared later, but never after the age of 10. Consanguinity and positive familial history were strongly associated in all groups. The ERG was extinct in LCA, showed reduced photopic response in achromatopsia and diffuse reduction in cone-rod distrophy. Color vision tests were altered mainly in achromatopsia and in cone distrophy. CONCLUSIONS: Retinal distrophies in childhood are a heterogeneous group of diseases with unspecific symptoms. A careful analysis of clinical features and diagnostic tests, specially color vision tests, ERG and visual field may be useful in their diagnosis.
Keywords: Vision; Photoreceptors; Color vision defects; Cones(Retina); Retinitis pigmentosa
INTRODUÇÃO
As distrofias retinianas são um grupo de doenças hereditárias que cursam com importante disfunção dos fotorreceptores (cones e/ou bastonetes). A distrofia mais conhecida e estudada é a retinose pigmentar, na qual os bastonetes são primariamente afetados, podendo ocorrer o acometimento mais tardio dos cones. Embora a retinose pigmentar possa se manifestar em praticamente todas as faixas etárias, a maioria dos pacientes começa a apresentar sintomas na adolescência e início da idade adulta.
As outras distrofias retinianas que se manifestam na infância são mais raras e, muitas vezes, de difícil diagnóstico.
A amaurose congênita de Leber (ACL) é a mais grave das distrofias retinianas com manifestação na infância, possuindo um padrão de herança autossômico recessivo. Caracteriza-se por baixa acuidade visual (BAV) severa manifestada logo ao nascimento ou no primeiro ano de vida(1). Inicialmente, o aspecto fundoscópico costuma ser normal, mas, com o passar do tempo, apesar de a acuidade visual se manter estável, podem surgir alterações, tais como dispersão difusa de pigmento, fundo "em sal e pimenta", atenuação das arteríolas, palidez do nervo óptico e, eventualmente, edema do disco óptico e alterações maculares que lembram um coloboma(2-3). A compressão óculo-digital (sinal de Franceschetti) costuma ser um sintoma marcante nestes pacientes e pode ser um fator importante no desenvolvimento de catarata e ceratocone na adolescência(4-5), além de ser responsável pelo enoftalmo comumente observado ainda na infância. Fazem parte, ainda, do quadro clínico o nistagmo, os reflexos pupilares lentos e a hipermetropia, muitas vezes, acima de +6,00 D. Alguns autores sugerem que a alta hipermetropia seja incluída como critério diagnóstico para um subtipo da ACL(6-7). O eletrorretinograma (ERG) comumente se mostra extinto ou quase extinto.
A ACL pode estar associada a vários quadros sistêmicos, principalmente ao retardo mental(1,8), que pode decorrer de alterações do sistema nervoso central ou pode ser conseqüência da privação dos estímulos visuais em períodos críticos da maturação nervosa(1). Têm sido relatadas associações com alterações neurológicas (hipoplasia cerebelar), cardiomiopatia(9), anormalidades esqueléticas, e características autísticas(10). Vários autores descrevem achados típicos da ACL em síndromes genéticas (síndrome de Joubert(11), síndrome de Senior-Loken, síndrome de Saldino-Mainzer) e doenças metabólicas.
As distrofias de cones são divididas em forma estacionária e forma progressiva(12). A forma estacionária, também conhecida como acromatopsia, se caracteriza pela ausência de função dos cones já ao nascimento. Na acromatopsia típica completa, os cones estão ausentes ou mal-formados, portanto a visão é fornecida apenas pelos bastonetes, daí o fato de também ser chamada de monocromatismo de bastonetes. Os pacientes são virtualmente cegos para cores, sendo capazes de perceber apenas diferenças de luminosidade. Geralmente, apresentam, já na primeira infância, nistagmo pendular, que tende a reduzir com a idade, baixa acuidade visual e marcante fotofobia. É bastante comum encontrarem-se altos graus de hipermetropia também nestes pacientes(12). A acuidade visual (AV) geralmente se situa em torno de 20/200, mas costuma melhorar em ambientes menos iluminados(12-13). A fundoscopia geralmente é normal ou mostra mínimas alterações, como redução do reflexo foveal ou finas granulações na mácula. O ERG mostra ausência de resposta dos cones (fase fotópica) e resposta normal para os bastonetes (fase escotópica). Nos testes de visão de cores, os pacientes conseguem reconhecer apenas a primeira prancha de Ishihara e mostram importantes alterações no panel D-15, geralmente sem eixo definido ou com um eixo de confusão que se situa entre o deutan e o tritan(14). O campo visual (CV) pode estar normal ou mostrar leve constrição. Um escotoma central às vezes pode ser demonstrado nos pacientes mais velhos. Existe uma forma incompleta da acromatopsia típica, onde há alguma função residual dos cones e os pacientes conseguem ter alguma discriminação para as cores. As duas formas, completa e incompleta, provavelmente são expressões diferentes de um mesmo genótipo e refletem o número de cones ausentes ou anormais(13). A acromatopsia típica tem herança autossômica recessiva. Já a forma atípica possui herança recessiva ligada ao X e também é conhecida como monocromatismo do cone azul, pois o sistema de cones S, responsável pela percepção do azul, é normal. Ela afeta, praticamente, só homens e seus sintomas tendem a ser menos severos. A AV normalmente se situa entre 20/80 e 20/200 e a maioria dos pacientes é míope. Nos testes de visão de cores, tendem a mostrar um eixo de confusão no deutan ou no protan, com menos erros no tritan(12).
As formas progressivas das distrofias de cones, por sua vez, parecem ser um grupo bastante heterogêneo de doenças, para as quais existem vários tipos de classificação e diferentes formas de herança(15). Elas se caracterizam pela deterioração precoce e progressiva da AV e da visão de cores, além de nistagmo e fotofobia. Procura-se fazer uma distinção entre distrofia de cones e distrofia de cones e bastonetes (ou mista). Na distrofia de cones pura, teoricamente, os bastonetes estariam normais. Já na distrofia mista haveria uma disfunção concomitante, ainda que menos severa, dos bastonetes. Porém, em muitos pacientes descritos como portadores de distrofia de cones, a função dos bastonetes é normal nos estágios iniciais, mas se deteriora à medida que a doença progride(12). Por isto alguns autores acreditam que a diferenciação não deveria ser feita e que todos os casos deveriam ser considerados como distrofia de cones e bastonetes. A distrofia de cones e a distrofia mista podem ter herança autossômica dominante (a mais comum), autossômica recessiva, recessiva ligada ao X(16-17) ou podem se manifestar de forma isolada. Geralmente observa-se uma redução gradual da AV na primeira e segunda décadas de vida, acompanhada de fotofobia e perda gradativa da visão de cores. A AV costuma ser pior durante o dia (hemeralopia), mas com a progressão da doença, torna-se ruim também à noite (nictalopia)(13). Nos estágios iniciais, a fundoscopia costuma ser normal ou apenas levemente alterada, com perda do reflexo foveal ou granulações na mácula. Posteriormente, pode-se observar uma atrofia oval do epitélio pigmentar da mácula, com aspecto de "bronze batido", às vezes associada à atrofia retiniana mais difusa. Uma maculopatia em "olho de boi" ("bull's eye") é freqüentemente notada, com importante simetria em ambos os olhos(13). Em estágios ainda mais avançados, surgem a palidez do disco óptico, o afilamento arteriolar difuso e a pigmentação periférica da retina. Os defeitos do campo visual podem incluir escotoma central, escotoma em anel, perda do CV periférico e redução difusa da sensibilidade. O ERG é normal nos estágios iniciais, evolui com redução da função dos cones e preservação da função dos bastonetes, até que, em estágios mais avançados, torna-se subvoltado de forma difusa, revelando o comprometimento também dos bastonetes(18-19).
Devemos citar também a doença de Stargardt, que geralmente se manifesta na primeira ou segunda décadas de vida e se caracteriza por piora progressiva da AV e alterações pigmentares na mácula, possuindo caráter autossômico recessivo.
Recentemente, foram realizadas, em nosso serviço, revisões dos casos de retinose pigmentar(20) e doença de Stargardt(21). Por esta razão, e por existirem poucos dados sobre o assunto na literatura nacional, decidimos realizar, no presente estudo, a análise dos casos das demais distrofias que se manifestam na infância: a ACL, a acromatopsia, a distrofia de cones e a distrofia mista. Procuramos traçar um perfil do quadro clínico e das alterações encontradas nos exames complementares de todos os pacientes portadores destas doenças atendidos no Serviço de Visão Subnormal do Hospital São Geraldo, Hospital das Clínicas da Universidade Federal de Minas Gerais, desde sua criação em 1992 até 2003.
MÉTODOS
Foram revistos os prontuários dos pacientes atendidos no Serviço de Visão Subnormal do Hospital São Geraldo, Hospital das Clínicas da UFMG, portadores de amaurose congênita de Leber (ACL), acromatopsia, distrofia de cones e distrofia mista, no período de 1992 a 2003. Selecionaram-se, também, os prontuários de pacientes que possuíam os diagnósticos iniciais de nistagmo congênito, BAV a esclarecer e distrofia a esclarecer, pois muitos destes pacientes poderiam ter sido posteriormente reclassificados.
Foram selecionados 51 prontuários, 11 dos quais não possuíam dados suficientes para que se pudesse chegar a uma conclusão diagnóstica. Permaneceram para análise os prontuários de 40 pacientes. Em alguns casos, após estudo cuidadoso do prontuário e dos exames complementares, foi feita a reclassificação do diagnóstico, dividindo-se os pacientes em quatro grupos: ACL, acromatopsia, distrofia de cones e distrofia mista.
Para a ACL, consideramos, como critérios diagnósticos, a baixa acuidade visual (BAV) severa ao nascimento ou no primeiro ano de vida, a fundoscopia inicial normal ou levemente alterada, a freqüente associação com a alta hipermetropia (>+6,00 D), e a presença do eletrorretinograma (ERG) extinto ou quase extinto.
Os critérios diagnósticos para a acromatopsia foram a BAV não progressiva, presente desde o nascimento ou ao primeiro ano de vida, a dificuldade na discriminação de cores, a fundosco pia normal ou levemente alterada, a freqüente associação com fotofobia e a presença do ERG com redução da resposta fotópica (cones) e preservação da resposta escotópica (bastonetes).
Os critérios diagnósticos para a distrofia de cones foram a BAV progressiva manifestada na infância, a dificuldade na discriminação de cores, a presença de alterações fundoscópicas, principalmente maculares, a freqüente associação com fotofobia e o ERG com redução da resposta fotópica e preservação da resposta escotópica.
Os principais dados que auxiliaram na diferenciação diagnóstica entre acromatopsia e distrofia de cones foram a piora progressiva da AV e a presença de alterações fundoscópicas características na distrofia de cones.
Os critérios diagnósticos para a distrofia mista foram os mesmos utilizados para a distrofia de cones, porém com o ERG mostrando disfunção difusa dos fotorreceptores, tanto na fase fotópica, quanto na escotópica.
Nem sempre foi possível preencher todos os critérios diagnósticos de cada patologia e, nestes casos, os pacientes foram classificados de acordo com os dados existentes no prontuário. Foram coletados dados referentes ao sexo, à idade na primeira consulta, diagnóstico inicial e final, sintomas e idade de seu aparecimento, história familiar, dados do exame oftalmológico (melhor AV com correção, refração, biomicroscopia, tonometria e fundoscopia) e exames complementares realizados. Chamamos de "consangüinidade" o relato de casamentos consangüíneos na família e de "história familiar positiva" a presença de outros membros da família com quadro semelhante.
Os testes de visão de cores relatados foram o de Ishihara, o Panel D-15, o Panel D-28 e o City University Color Vision Test, sendo que vários pacientes foram submetidos a todos. O campo visual, quando realizado, foi o de Goldmann.
Os dados foram analisados através do programa Epi-Info 2000, versão 1.1.2.
RESULTADOS
Os dados referentes ao sexo, à idade na primeira consulta, acuidade visual, refração estática e fundoscopia estão agrupados na tabela 1. Encontramos 10 pacientes portadores de ACL, 17 com acromatopsia, 6 com distrofia de cones e 7 com distrofia mista.
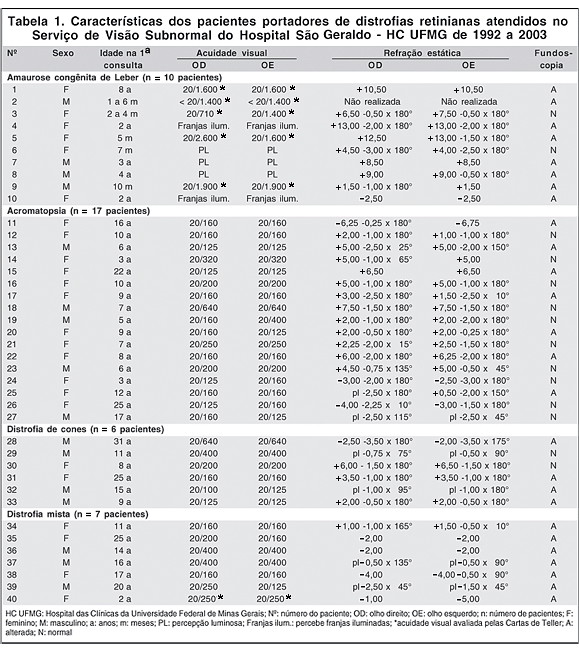
Da amostra, 24 pacientes (60%) eram do sexo feminino e 16 (40%) do sexo masculino. No grupo da ACL, 6 pacientes (60%) eram do sexo feminino e 4 (40%) do sexo masculino. Na acromatopsia, 12 (70,6%) eram do sexo feminino e 5 (29,4%) do sexo masculino. Na distrofia de cones, 2 (33,3%) eram do sexo feminino e 4 (66,6%) do sexo masculino e no grupo da distrofia mista, 4 (57,1%) eram do sexo feminino e 3 (42,9%) do sexo masculino.
No grupo da ACL, a idade na primeira consulta variou de 5 meses a 8 anos, com média de 2,5 e mediana de 2 anos. A acuidade visual variou de 20/710 a percepção luminosa e a refração variou de -2,50 D a +13,00 -1,50 x 180º. Seis pacientes (60%) eram portadores de alta hipermetropia, 2 (20%) eram hipermétropes, 1 (10%) era míope e, em 1 paciente (10%), a refração era desconhecida. A fundoscopia no primeiro exame se mostrou normal em 2 pacientes (20%) e alterada em 8 pacientes (80%). As alterações relatadas foram redução do reflexo foveal, mosqueamento e rarefação difusa do epitélio pigmentar da retina (EPR), fundo em sal e pimenta, mácula com aspecto de cobre batido, "displasia" da mácula, manchas brancas ou lesões amareladas e arredondadas na média periferia, palidez da papila, pseudopapiledema e afilamento arteriolar. A paciente nº 6, acompanhada dos 7 meses aos 8 anos, apresentava fundoscopia normal no primeiro ano de vida, aos 6 anos já apresentava afilamento arteriolar e, aos 8 anos, mostrava fundo em sal e pimenta, redução do reflexo foveal e pseudopapiledema em um dos olhos, que, com a investigação, revelou tratar-se de drusas do nervo óptico. A paciente nº 4, portadora de síndrome de Joubert, apresentava grave retinopatia hipertensiva, devido ao comprometimento renal.
No grupo da acromatopsia, a idade na primeira consulta variou de 3 a 25 anos, com média de 10,3 e mediana de 9 anos. A acuidade visual variou de 20/125 a 20/640, com média em 20/200. A refração variou de -6,75 D a +7,50 -1,50 x 180º. Três pacientes (17,6%) eram portadores de alta hipermetropia, 9 (52,9%) eram hipermétropes, 3 (17,6%) eram míopes e 2 (11,8%) possuíam astigmatismo. A fundoscopia no primeiro exame se mostrou normal em 10 pacientes (58,8%) e alterada em 7 (41,2%). As alterações descritas foram discreto ou moderado afilamento arteriolar, redução do reflexo foveal, alterações pigmentares na mácula, e discreta palidez papilar.
No grupo da distrofia de cones, a idade na primeira consulta variou de 8 a 31 anos, com média de 16,5 e mediana de 13 anos. A acuidade visual variou de 20/125 a 20/640, com média de 20/280. A refração variou de -2,50 -3,50 x 180º a +6,00 -1,50 x 180º. Um paciente (16,7%) era alto hipermétrope, 2 (33,3%) eram hipermétropes, 1 (16,6%) era míope e 2 (33,3%) possuíam astigmatismo. A fundoscopia no primeiro exame se mostrou normal em 2 pacientes (33,3%) e alterada em 4 (66,7%). As alterações descritas foram maculopatia em "olho de boi" e rarefação e dispersão de pigmentos na mácula. O paciente nº 30, que apresentava fundoscopia inicial normal aos 8 anos, evoluiu, até os 12 anos, com perda do reflexo foveal, afilamento vascular difuso e pigmentação esparsa periférica.
No grupo da distrofia mista, a idade na primeira consulta variou de 2 a 25 anos, com média de 15 e mediana de 16 anos. A acuidade visual variou de 20/160 a 20/400, com média em 20/260. A refração variou de -5,00 D a +1,50 -1,00 x 165º. Um paciente (14,3%) era hipermétrope, 4 (57,1%) eram míopes e 2 (28,6%) possuíam astigmatismo. A fundoscopia se mostrou alterada em todos os pacientes (100%), tendo sido relatadas alterações pigmentares generalizadas, palidez da papila, redução do reflexo macular, mácula com aspecto granular ou de metal batido e afilamento arteriolar,
Os sintomas dos pacientes estão sumarizados na tabela 2. A baixa acuidade visual foi relatada em todos os pacientes (100% dos casos).
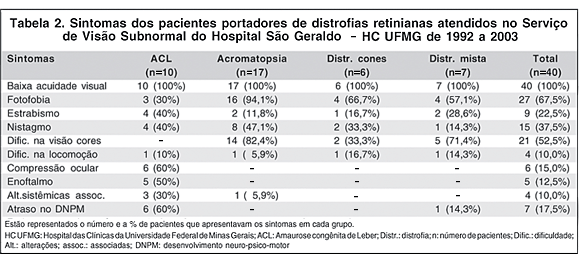
No grupo da ACL, além da BAV, os sintomas mais freqüentes foram a compressão óculo-digital (6 pacientes, 60%), o enoftalmo (5 pacientes, 50%), o estrabismo e o nistagmo (4 pacientes cada, 40%). A fotofobia foi relatada em 3 pacientes (30%) e a dificuldade na locomoção em 1 (10%). Foram encontradas alterações sistêmicas em 3 pacientes (30%), sendo que uma era portadora da síndrome de Joubert, outra possivelmente possuía a síndrome de Saldino-Mainzer e, em outro, as alterações sistêmicas não foram discriminadas. O atraso no desenvolvimento neuro-psico-motor (DNPM) ocorreu em 6 pacientes (60%).
No grupo da acromatopsia, além da BAV, os sintomas mais freqüentes foram a fotofobia (16 pacientes, 94,1%), a dificuldade na visão de cores (14 pacientes, 82,4%) e o nistagmo (8 pacientes, 47,1%). Menos freqüentes foram o estrabismo (2 pacientes, 11,8%) e a dificuldade na locomoção (1 paciente, 5,9%). A associação com alterações sistêmicas foi descrita em apenas 1 paciente (5,9%), o de nº 23, portador de síndrome de Ehlers-Danlos.
No grupo da distrofia de cones, além da BAV, os sintomas mais freqüentes foram a fotofobia (4 pacientes, 66,7%), a dificuldade na visão de cores (2 pacientes, 33,3%) e o nistagmo (2 pacientes, 33,3%). Um paciente (16,7%) apresentava estrabismo e 1 (16,7%) relatava dificuldade na locomoção. Não foram encontradas alterações sistêmicas.
No grupo da distrofia mista, além da BAV, os sintomas mais freqüentes foram a dificuldade na visão de cores (5 pacientes, 71,4%) e a fotofobia (4 pacientes, 57,1%). Menos freqüentes foram o estrabismo (2 pacientes, 28,6%), o nistagmo e a dificuldade na locomoção (1 paciente cada, 14,3%). Não foram encontradas alterações sistêmicas, mas 1 paciente (14,3%) apresentava atraso no DNPM.
A época da observação dos sintomas está relatada na tabela 3. Todos os pacientes apresentaram sintomas no máximo até os 10 anos, sendo que 20 (50%) já os tinham ao nascimento e 13 (32,5%) os apresentaram no primeiro ano de vida. Apenas 7 pacientes (17,5%) manifestaram os primeiros sintomas após 1 ano de vida, sendo 4 (10%) entre 1 e 3 anos, 1 (2,5%) entre 4 e 6 anos e 2 (5%) entre 7 e 10 anos.
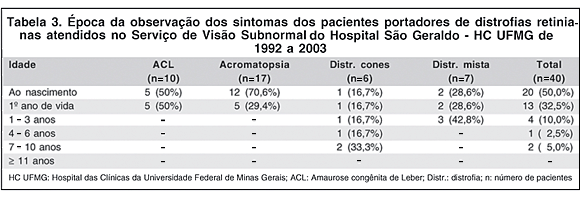
Na ACL, 5 pacientes (50%) manifestaram os sintomas ao nascimento e 5 (50%) durante o primeiro ano de vida. Na acromatopsia, 12 pacientes (70,6%) já tinham sintomas ao nascimento e 5 (29,4%) no primeiro ano de vida. Tanto na ACL quanto na acromatopsia, não houve casos com início dos sintomas após 1 ano de idade. Já na distrofia de cones, encontramos 1 paciente (16,7%) com sintomas ao nascimento, 1 (16,7%) no primeiro ano de vida, 1 (16,7%) entre 1 e 3 anos, 1 (16,7%) entre 4 e 6 anos e 2 (33,3%) entre 7 e 10 anos, enquanto que, na distrofia mista, 2 pacientes (28,6%) manifestaram os sintomas ao nascimento, 2 (28,6%) no primeiro ano de vida e 3 (42,8%) entre 1 e 3 anos.
Os diagnósticos iniciais feitos no encaminhamento dos pacientes ao Serviço de Visão Subnormal estão listados na tabela 4. O diagnóstico inicial mais freqüente foi o nistagmo congênito (11 pacientes, 27,5%), seguido de distrofia a esclarecer (8 casos, 20%).
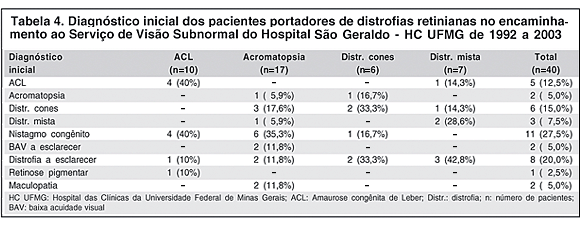
No grupo da ACL, 4 pacientes (40%) vieram encaminhados como ACL, 4 (40%) como nistagmo congênito, 1 (10%) como distrofia a esclarecer e 1 (10%) como retinose pigmentar.
Na acromatopsia, apenas 1 paciente (5,9%) veio encaminhado com o diagnóstico de acromatopsia. Seis pacientes (35,3%) foram inicialmente diagnosticados como nistagmo congênito, 3 (17,6%) como distrofia de cones, 2 (11,8%) como BAV a esclarecer, 2 (11,8%) como distrofia a esclarecer, 2 (11,8%) como maculopatia e 1 (5,9%) como distrofia mista.
Na distrofia de cones, 2 pacientes (33,3%) vieram encaminhados como distrofia de cones, 2 (33,3%) como distrofia a esclarecer, 1 (16,7%) como acromatopsia e 1 (16,7%) como nistagmo congênito.
Na distrofia mista, 2 pacientes (28,6%) vieram encaminhados como portadores de distrofia mista, 3 (42,8%) como distrofia a esclarecer, 1 (14,3%) como ACL e 1 (14,3%) como distrofia de cones.
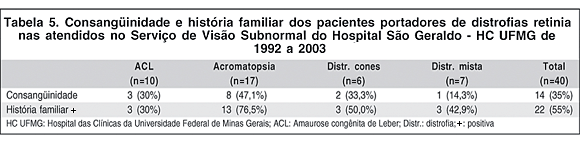
A consangüinidade e a história familiar estão resumidas na tabela 5. Da amostra, 14 pacientes (35%) relatavam consangüinidade na família e 22 (55%) tinham história familiar positiva, ou seja, descreviam outros familiares com quadros semelhantes. No grupo da ACL, 3 pacientes (30%) relataram consangüinidade e 3 (30%) história familiar. Na acromatopsia, 8 (47,1%) tinham consangüinidade e 13 (76,5%) história familiar. Na distrofia de cones 2 (33,3%) tinham consangüinidade e 3 (50%) história familiar, enquanto que, na distrofia mista, 1 (14,3%) relatava consangüinidade e 3 (42,9%) história familiar.
Os exames complementares estão agrupados na tabela 6, que descreve o eletrorretinograma (ERG), os teste de visão de cores e o campo visual (CV).
Da amostra, 24 pacientes (60%) realizaram o ERG, que se mostrou extinto apenas nos portadores de ACL, com redução difusa apenas nos portadores de distrofia mista e com redução da resposta fotópica nos casos de acromatopsia e distrofia de cones. Na ACL, 7 pacientes (70%) tinham o ERG extinto e 3 (30%) não o realizaram. Na acromatopsia, 6 (35,3%) mostraram redução da resposta fotópica e 11 (64,7%) não o fizeram. Na distrofia de cones, 4 (66,7%) tinham redução da resposta fotópica e 2 (33,3%) não o realizaram, enquanto que, na distrofia mista, todos os 7 (100%) mostravam redução difusa no ERG.
Os testes de visão de cores foram realizados em 18 pacientes (45%). Na ACL, os testes não foram feitos em 9 casos (90%) e apenas 1 paciente (10%) mostrou uma tritanopia. Na acromatopsia, 10 pacientes (58,9%) mostraram alterações sem eixo definido, 1 (5,9%) mostrou alterações sugestivas de protanopia e 1 (5,9%) de tritanopia, enquanto que 5 (29,4%) não fizeram os testes. Na distrofia de cones, 1 paciente (16,7%) tinha protanopia, 1 (16,7%) tinha tritanopia, 1 (16,7%) tinha alterações sem eixo definido e 3 (50%) não fizeram os testes. Na distrofia mista, 1 paciente (14,3%) tinha a visão de cores normal, 2 (28,6%) tinham alterações sem eixo definido e 4 (57,1%) não fizeram os testes.
O campo visual foi realizado em apenas 16 pacientes (40%). No grupo da ACL, apenas 1 paciente (10%) mostrou uma contração importante de todas as isópteras, com campo tubular restrito aos 10º centrais em ambos os olhos, enquanto que, nos outros 9 pacientes (90%) o exame nem foi solicitado. Na acromatopsia, o CV foi normal em 1 caso (5,9%), mostrou contração difusa em 9 (52,9%) e não foi realizado em 7 (41,2%). Na distrofia de cones o CV mostrou contração difusa em 1 paciente (16,7%), escotoma central em 1 (16,7%) e não foi realizado em 4 (66,6%), enquanto que, na distrofia mista, mostrou contração difusa em 1 caso (14,3%), escotoma central em 2 (28,6%) não foi realizado em 4 pacientes (57,1%).
DISCUSSÃO
As distrofias retinianas que se manifestam na infância são um grupo heterogêneo de doenças hereditárias que cursam com disfunção dos fotorreceptores e, por serem raras e apresentarem sintomas não específicos, representam um desafio para seu diagnóstico.
A amaurose congênita de Leber (ACL) é a mais grave de todas as distrofias da infância e se caracteriza por BAV severa ao nascimento ou no primeiro ano de vida, fundoscopia inicialmente normal ou com leves alterações e ERG extinto ou quase extinto(1), podendo ocorrer como uma doença ocular isolada ou associada a síndromes genéticas e doenças metabólicas(3,9,11,22). A acromatopsia é uma distrofia também congênita e se caracteriza pela ausência da função dos cones(12-14). Seus sintomas costumam se manifestar ao nascimento ou no primeiro ano de vida, cursando com BAV estável e menos severa do que na ACL, incapacidade para a discriminação de cores, nistagmo, fotofobia importante e fundoscopia normal ou com leves alterações. O ERG, caracteristicamente mostra ausência de resposta fotópica (mediada pelos cones) e resposta escotópica (mediada pelos bastonetes) normal. A distrofia de cones apresenta sintomas bastante semelhantes aos da acromatopsia, porém estes são progressivos e podem se manifestar em idades mais avançadas. Aqui, a fundoscopia já costuma mostrar alterações bem evidentes, principalmente maculares, também progressivas, e o ERG mostra, inicialmente, disfunção dos cones, mas pode evoluir com acometimento também dos bastonetes nas fases mais avançadas da doença. A distrofia mista costuma ter sintomas parecidos com os da distrofia de cones, porém o ERG mostra disfunção tanto dos bastonetes quanto dos cones. Alguns autores consideram a distrofia mista um estágio evolutivo mais avançado da distrofia de cones(12-13).
Em nosso estudo tivemos a oportunidade de observar as semelhanças e as diferenças entre os quatro grupos de doenças estudadas.
A idade dos pacientes na primeira consulta costuma refletir a época em que os pais buscam ajuda para os sintomas do paciente. No grupo da ACL, ela se situou em torno dos 2 anos, sendo que o paciente mais jovem tinha 5 meses. A busca por ajuda tão cedo possivelmente está relacionada à gravidade e à precocidade dos sintomas neste grupo. O paciente mais velho, de 8 anos, nos foi encaminhado por uma instituição para cegos, onde já vinha sendo acompanhado. No grupo da acromatopsia, apesar de todos os pacientes já terem manifestado os sintomas até o primeiro ano de vida, a mediana das idades na primeira consulta foi de 9 anos. A maioria dos pacientes já estava em idade escolar ou produtiva, época em que, talvez, seus sintomas tenham começado a interferir com as atividades da escola ou do trabalho. Na distrofia de cones e na distrofia mista, a mediana das idades foi de 13 e 16 anos, respectivamente, e, neste caso, pode refletir tanto o aparecimento mais tardio dos sintomas quanto à época em que eles começaram a interferir com as atividades dos pacientes.
A baixa acuidade visual esteve presente em todos os pacientes do estudo, sendo muito mais grave na ACL, onde todos os pacientes seriam classificados como portadores de visão subnormal profunda ou quase cegueira. A BAV severa na ACL é relatada em todos os trabalhos da literatura. Ainda que alguns autores relatem portadores de ACL com AV tão boa quanto 20/100(2), até casos de ausência de percepção luminosa(1-2,22), em nossa amostra, a melhor AV foi de 20/710 e nenhum dos pacientes era totalmente cego. Nos outros grupos, a AV média foi melhor, sendo de 20/200 na acromatopsia, 20/280 na distrofia de cones e 20/260 na distrofia mista. Quase todos os pacientes seriam classificados como portadores de visão subnormal moderada ou severa. Nossos achados coincidem com os da literatura(12-13).
Os erros refracionais foram extremamente prevalentes em nossa amostra. Chama a atenção a grande freqüência da alta hipermetropia (60% dos casos) e da hipermetropia (20%) na ACL. Vários autores relatam esta associação(6-7,9,23), sendo que um deles(6) chega a sugerir que a alta hipermetropia seja incluída como critério diagnóstico para um subtipo da ACL. A única paciente míope era uma criança possivelmente com a síndrome de Saldino-Mainzer. Também no grupo da acromatopsia verificamos grande prevalência de hipermétropes (52,9%) e altos hipermétropes (17,6%), dado igualmente encontrado na literatura(12). Na distrofia de cones a hipermetropia também predominou (50%), enquanto que, na mista, a miopia foi o principal erro refracional (57,1%).
A fundoscopia tem sido descrita como normal ou minimamente alterada nos estágios iniciais da ACL, havendo posterior desenvolvimento de alterações de aspecto extremamente variável(1-4,22). Em nossa amostra, a maioria dos pacientes (80%) já apresentava alterações fundoscópicas no primeiro exame, talvez por este ter sido realizado em épocas em que aquelas já tinham se tornado evidentes. Encontramos descrições clássicas e concordantes com a literatura, tais como a rarefação difusa do EPR, fundo em sal e pimenta, palidez da papila, pseudopapiledema e afilamento arteriolar. Em uma das pacientes com fundoscopia normal, foi possível observar o aparecimento gradativo de alterações típicas (fundo em sal e pimenta e afilamento vascular) e, até mesmo, de um achado nunca antes relatado na ACL, as drusas do nervo óptico. Na acromatopsia, a fundoscopia é descrita como normal ou levemente alterada(12-13), e estável. Em nossa amostra, a maioria dos pacientes (58,8%) tinha aspecto fundoscópico normal. Nos pacientes onde foram relatadas alterações, estas foram predominantemente leves, como afilamento arteriolar, discreta palidez da papila, redução do reflexo foveal e alterações pigmentares na mácula. Na distrofia de cones, a fundoscopia, inicialmente normal, evolui com alterações maculares progressivas, que, em estágios mais avançados, podem atingir a periferia(12-13,15). No presente estudo, 2 dos 6 pacientes tinham fundoscopia inicial normal, sendo que um deles evoluiu posteriormente com perda do reflexo foveal, afilamento vascular difuso e pigmentação esparsa periférica. Alterações predominantemente maculares foram descritas em 5 dos 6 pacientes, sendo que, em um deles, foi encontrada a clássica maculopatia em "olho de boi"(12). Por fim, na distrofia mista, a fundoscopia geralmente está alterada(12-13), exatamente o que foi observado em nossa amostra (100% dos casos).
Quanto aos sintomas, a compressão óculo-digital e o enoftalmo foram exclusivos da ACL, que também mostrou grande freqüência de estrabismo e nistagmo. Apesar de não ser um sintoma típico, alguns trabalhos mostram a presença de fotofobia na ACL(3-4,8), o que pôde ser verificado em 30% de nossa amostra. Nas outras três patologias, a fotofobia e a dificuldade na discriminação de cores foram os sintomas mais prevalentes, principalmente na acromatopsia(23). Estes dados podem ser de grande auxílio no diagnóstico destas doenças.
O retardo no desenvolvimento neuro-psico-motor (DNPM) tem sido freqüentemente descrito em associação com a ACL(1,8). Questiona-se se é devido a alterações neurológicas associadas ou se decorre apenas da privação sensorial imposta pela doença. Em nossa amostra, ele foi muito freqüente, ocorrendo em 60% dos casos. A ACL também tem sido descrita em associação com algumas síndromes genéticas(8-9,11,22), como a síndrome de Joubert (hipoplasia cerebelar e problemas respiratórios), a síndrome de Senior-Loken (nefronophthisis), e a síndrome de Saldino-Mainzer (displasia esquelética, nefrophthisis e ataxia cerebelar). Em nossa amostra, uma paciente tinha a síndrome de Joubert e outra, possivelmente, a de Saldino-Mainzer, hipótese levantada durante a revisão do prontuário. Nas outras distrofias, apenas um paciente acromata possuía alterações sistêmicas associadas, no caso a síndrome de Ehlers-Danlos. Não conseguimos encontrar esta associação na literatura pesquisada.
Outro instrumento importante no diagnóstico diferencial é a pesquisa da época de aparecimento dos sintomas. Na ACL e na acromatopsia, doenças sabidamente congênitas, todos os pacientes apresentaram os sintomas ao nascimento ou no primeiro ano de vida, o que está em perfeito acordo com os dados da literatura(1-3,12-13). Já na distrofia de cones e na distrofia mista, apesar de alguns pacientes já manifestarem os sintomas ao nascimento, muitos iniciaram sua sintomatologia um pouco mais tarde, o que também confere com a literatura(12-13,24). Nenhum dos quadros, no entanto, teve início após os 10 anos de idade.
Pelo fato de apresentarem sintomas variados e pouco específicos, as distrofias retinianas podem ser de difícil diagnóstico diferencial, o que pode ser claramente observado quando se analisam os diagnósticos iniciais de nossos pacientes. A grande maioria nos foi encaminhada com o diagnóstico de nistagmo congênito ou de distrofia a esclarecer. Somente após a coleta de uma história detalhada e a realização de exames complementares, principalmente o ERG, os pacientes puderam ser corretamente classificados. Ainda assim, muitos diagnósticos ficaram sem confirmação final, pois alguns pacientes não tiveram acesso a exames complementares essenciais ou simplesmente não retornaram. Acreditamos que este seja um importante fator de confusão em nosso estudo e que alguns pacientes ainda estejam classificados de forma incorreta. Pudemos observar também a grande dificuldade na diferenciação entre a acromatopsia e a distrofia de cones devido à semelhança de seus sintomas e de suas alterações nos exames complementares. Por esta razão, todos os pacientes com estes diagnósticos estão sendo atualmente chamados para serem novamente estudados.
Alguns autores(25-26) ressaltam a importância da investigação de uma criança com nistagmo congênito, pois o nistagmo é um sintoma inespecífico de inúmeras patologias, dentre elas as distrofias retinianas. Nestes casos, o ERG é de extrema valia para o diagnóstico diferencial.
As distrofias retinianas possuem um padrão de herança razoavelmente estabelecido. A ACL e a acromatopsia típica são autossômicas recessivas, a acromatopsia atípica é recessiva ligada ao X e as distrofias de cones e mista costumam ser autossômicas dominantes(27), apesar de existirem as formas autossômica recessiva, recessiva ligada ao X(16-17) e a isolada. A alta prevalência da consangüinidade e da história familiar em todos os grupos do presente estudo confirma o caráter heredo-familiar destas distrofias retinianas.
Os exames complementares são essenciais para o diagnóstico das distrofias, principalmente se a fundoscopia é normal. Os principais são o ERG, os testes de visão de cores e o campo visual. Infelizmente uma grande porcentagem de nossa amostra não pôde realizá-los, devido a sua pouca disponibilidade na época, ou à dificuldade de sua aplicação em pacientes muito jovens. Assim, alguns diagnósticos tiveram de ser realizados tendo como base apenas a história e os achados clínicos, o que pode reduzir sua confiabilidade.
Nos pacientes em que o ERG foi realizado, ele se mostrou extinto na ACL, com redução da resposta fotópica na acromatopsia e na distrofia de cones e difusamente subvoltado na distrofia mista, tendo certamente sido utilizado para o diagnóstico diferencial destes pacientes. Os testes de visão de cores mostraram importantes alterações principalmente na acromatopsia e na distrofia de cones. De uma forma geral, a principal alteração encontrada no CV foi a contração difusa das isópteras. A presença de um escotoma central pôde ser verificada em apenas 1 paciente da distrofia de cones e 2 da distrofia mista. A ausência de dados referentes aos testes de visão de cores e ao CV em grande parte da amostra torna difícil qualquer correlação entre os achados e as patologias.
CONCLUSÃO
As distrofias retinianas da infância são doenças raras e de difícil diagnóstico, podendo ter múltiplas manifestações clínicas e diferentes tipos de herança. Diante de uma criança com baixa visão, deve-se lançar mão de vários recursos diagnósticos, dentre eles a anamnese cuidadosa, o exame oftalmológico completo e os exames complementares (ERG, testes de visão de cores e campo visual). Ainda assim, em alguns casos, apenas o tempo poderá levar ao diagnóstico correto, mostrando a evolução da doença. Um diagnóstico bem feito determinará o prognóstico e auxiliará no aconselhamento aos pais e na conduta frente a cada paciente.
REFERÊNCIAS
1. Nickel B. Hoyt CS. Leber's congenital amaurosis. Is mental retardation a frequent associated defect? Arch Ophthalmol. 1982;100(7):1089-92.
2. Heher KL, Traboulsi EI, Maumenee IH. The natural history of Leber's congenital amaurosis. Age-related findings in 35 patients. Ophthalmology. 1992;99(2):241-5.
3. Schroeder R, Mets MB, Maumenee IH. Leber's congenital amaurosis. Retrospective review of 43 cases and a new fundus finding in two cases. Arch Ophthalmol. 1987;105(3):356-9.
4. Smith D, Oestreicher J, Musarella MA. Clinical spectrum of Leber's congenital amaurosis in the second to fourth decades of life. Ophthalmology. 1990;97 (9):1156-61.
5. Waisberg Y, Katina JH. Ceratocone associado à cegueira congênita e compressão digito-ocular. Arq Bras Oftalmol. 1989;52(1):28-30.
6. Wagner RS, Caputo AR, Nelson LB, Zanoni D. High hyperopia in Leber's congenital amaurosis. Arch Ophthalmol. 1985;103(10):1507-9.
7. Babel J, Klein D, Roth A. Leber's congenital amaurosis associated with high hyperopia in four sisters. Ophthalmic Paediatr Genet. 1989;10(1):55-61.
8. Schuil J, Meire FM, Delleman JW. Mental retardation in amaurosis congenita of Leber. Neuropediatrics. 1998;29(6):294-7.
9. Russel-Eggitt IM, Taylor DS, Clayton PT, Garner A, Kriss A, Taylor JF. Leber's congenital amaurosis - a new syndrome with a cardiomyopathy. Br J Ophthalmol. 1989;73(4):250-4.
10. Rogers SJ, Newhart-Larson S. Characteristics of infantile autism in five children with Leber's congenital amaurosis. Dev Med Child Neurol. 1989;31(5): 598-608.
11. Ivarsson SA, Bjerre I, Brun A, Ljungberg O, Maly E, Taylor I. Joubert syndrome associated with Leber amaurosis and multicystic kidneys. Am J Med Genet. 1993;45(5):542-7.
12. Simunovic MP, Moore AT. The cone dystrophies. Eye. 1998;12(Pt 3b):553-65. Review.
13. Cavender JC, Ai E. Hereditary Macular Dystrophies. In: Duane TD, Jaeger EA. Clinical Ophthalmology. Philadelphia: Lippincott; 1988. Capítulo 9. p.1-29.
14. Haegerstrom-Portnoy G, Schneck ME, Verdon WA, Hewlett SE. Clinical vision characteristics of the congenital achromatopsias. II. Color vision. Optom Vis Sci. 1996;73(7):457-65.
15. François J, De Rouck A, De Laey JJ. Progressive cone dystrophies. Ophthalmologica. 1976;173(2):81-101.
16. Jacobson DM, Thompson HS, Bartley JA. X-linked progressive cone dystrophy. Clinical characteristics of affected males and female carriers. Ophthalmology. 1989;96(6):885-95.
17. Mantyjarvi M, Nurmenniemi P, Partanen J, Myohanen T, Peippo M, Alitalo T. Clinical features and a follow-up study in a family with X-linked progressive cone-rod dystrophy. Acta Ophthalmol Scand. 2001;79(4):359-65.
18. Mendonça RF, Takahashi WY, Usuba FS, Mendonça CHF. Alterações eletrorretinográficas na distrofia de cones. Rev Bras Oftalmol. 1999;58(1):21-5.
19. Sato M, Sacai PY, Berezovsk A, Salomão SR. Avaliação da função visual em pacientes com distrofia de cones. Arq Bras Oftalmol. 2003;66(3):293-7.
20. Moya STF, Oliveira AG, Fernandes LC. Retinose pigmentária e visão subnormal: adaptação aos auxílios ópticos para perto e correlação com funções visuais. Rev Bras Oftalmol. 2003;62(9):679-88.
21. Carvalho LC, Fernandes LC. Doença de Stargardt: Epidemiologia, Quadro clínico e utilização de auxílios em visão subnormal [monografia]. Belo Horizonte: Universidade Federal de Minas Gerais; 2003.
22. Lambert SR, Kriss A, Taylor D, Coffey R, Pembrey M. Follow-up and diagnostic reappraisal of 75 Patients with Leber's congenital amaurosis. Am J Ophthalmol. 1989; 107(6):624-31. Review.
23. Young RS, Price J, Harrison J. Aversion to daytime illumination in patients with congenital achromatopsia. Percept Mot Skills. 1987;64(3 Pt 1):923-6.
24. Sadowski B, Zrenner E. Cone and rod function in cone degenerations. Vision Res. 1997;37(16):2303-14.
25. Good PA, Searle AE, Campbell S, Crews SJ. Value of the ERG in congenital nystagmus. Br J Ophthalmol. 1989;73(7):512-5.
26. Lambert SR, Taylor D, Kriss A. The infant with nystagmus, normal appearing fundi, but an abnormal ERG. Surv Ophthalmol. 1989;34(3):173-86. Review.
27. Ripps H, Noble KG, Greenstein VC, Siegel IM, Carr RE. Progressive cone distrophy. Trans Am Ophthalmol Soc. 1987;85:82-100.
Endereço para correspondência
Heloisa Andrade Maestrini, Rua Maranhão 655 - Belo Horizonte (MG)
Cep 30150-330
E-mail: [email protected]
Recebido para publicação em 01.03.2004
Aprovação em 23.07.2004
Trabalho realizado no Serviço de Visão Subnormal do Hospital São Geraldo, Hospital das Clínicas da Universidade Federal de Minas Gerais - UFMG.
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos à Dra. Rosane da Cruz Ferreira.
© 2024 - All rights reserved - Conselho Brasileiro de Oftalmologia
![]()
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Scielo
Scielo
 Pocket
Pocket
