
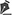
Mariana Matioli da Palma1,2; Daniel Martin1; Mariana Vallim Salles2,3; Fabiana Louise Teixeira Motta2,3; Suel Abujamra1; Juliana Maria Ferraz Sallum2,3
DOI: 10.5935/0004-2749.20190033
ABSTRACT
This report presents three patients diagnosed with macular dystrophies with variants in PRPH2. Peripherin-2, the protein of this gene, is important in the morphogenesis and stabilization of the photoreceptor outer segment. Peripherin-2 deficiencies cause cellular apoptosis. Moreover, pathogenic variants in PRPH2 are associated with various diseases, such as pattern, butterfly-shaped pattern, central areolar, adult-onset vitelliform macular, and cone-rod dystrophies as well as retinitis pigmentosa, retinitis punctata albescens, Leber congenital amaurosis, fundus flavimaculatus, and Stargardt disease.
Keywords: Retinal degeneration; Genotyping techniques; Vitelliform macular dystrophy; Retinitis pigmentosa; Genetics
RESUMO
Este relato apresenta três pacientes com diagnóstico de distrofias maculares com mutações no PRPH2. Periferina 2, a proteína deste gene, é importante na morfogênese e estabilização do segmento externo dos fotorreceptores. Deficiências de periferina 2 causam apoptose celular. Além disso, variantes patogênicas no PRPH2 estão relacionadas a diferentes doenças, como distrofia padrão, distrofia padrão em asa de borboleta, distrofia central areolar, distrofia viteliforme do adulto, retinose pigmentar, distrofia de cones e bastonetes, retinite punctata albscens, amaurose congênita de Leber, fundus flavimaculatus e doença de Stargardt.
Descritores: Degeneração retiniana; Técnicas de genotipagem; Distrofia macular viteliforme; Retinite pigmentosa; Genética
INTRODUCTION
PRPH2, previously known as RDS (retinal degeneration slow), was initially identified to cause natural retinal degeneration in rats(1). The peripherin-2 protein codified for this gene is essential in the morphogenesis of the photoreceptor outer segment composed of parallel, closely packed bilaminar discs in the plasma membrane. Each disc contains a rim and a lamella. The rim surrounds the extremity of the disc and maintains the flattened morphology; the lamella is where phototransduction occurs. Vision is affected when this structure is altered(2,3).
Peripherin-2 is a transmembrane glycoprotein that is intrinsic to both the curvature formation of each disc and the flattened surface morphology. Deficiency of this protein results in cellular disorganization and cellular apoptosis activation via unknown mechanisms(2,3).
Here, three patients are described with pathogenic variants in PRPH2 and their distinct clinical diagnoses.
CASE REPORTS
Patient 1 was a 43-year-old man presented with visual haze and photophobia. Best-corrected visual acuity (BCVA) was 20/80 in the right eye and 20/100 in the left eye, and he was diagnosed with cone dystrophy (CD). Funduscopy revealed atrophy of the retinal pigment epithelium in the macular area, whereas autofluorescence exhibited a pattern of extensive hypoautofluorescent marbleization in the posterior pole. Optical coherence tomography (OCT) revealed retinal atrophy with irregularities in the outermost layers and a disrupted ellipsoid layer (Figure 1A, 1B, 1C).
Sanger sequencing of PRPH2 revealed a pathogenic homozygous variant (p.Gly208Asp [c.623G>A]) and three benign homozygous variants (p.Gln304Glu [c.910C>G], p.Arg310Lys [c.929G>A], and p.Asp338Gly [c.1013A>G]).
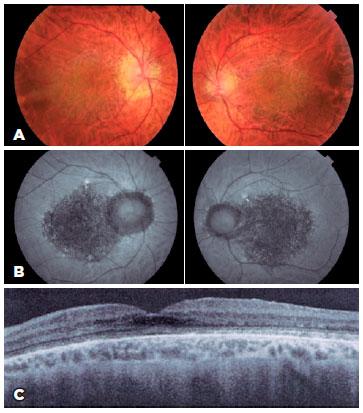
Patient 2 was a 50-year-old man with a 5-year history of blurred distance vision. BCVA was 20/40 in the right eye and 20/25 in the left eye, and he was diagnosed with adult-onset vitelliform macular dystrophy (AVMD). The retinal map revealed a macular vitelliform lesion, and angiofluoresceinography revealed macular hyperfluorescence due to window defect without contrast extravasation. In addition, a juxtafoveal hyper-reflective vitelliform lesion with diffuse thinning of the macular area was identified on OCT (Figure 2A, 2B, 2C).
A pathogenic heterozygous variant p.Ile33Hisfs*12 (c.96dupC) was found in PRPH2 using panel-based sequencing for macular dystrophy.
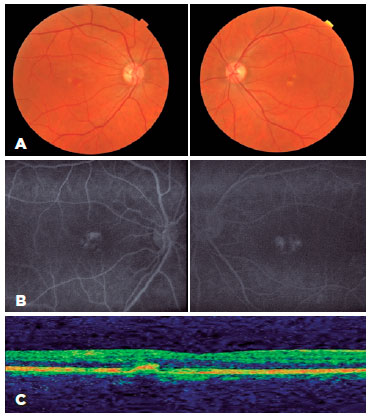
Patient 3 was a 59-year-old woman with visual haze since infancy. Her bilateral BCVA was 20/400. She was diagnosed with Stargardt disease; her sister, father, paternal aunt, and paternal great uncle, too, were similarly affected. Macular atrophy was revealed on funduscopy. Fundus autofluorescence revealed a hypoautofluorescent central lesion corresponding to macular atrophy contrasting with a hyperautofluorescent peripheral lesion. OCT revealed diffuse thinning of all retinal layers with diminished foveal depression and atrophy of the external layer (Figure 3A, 3B, 3C).
Using panel sequencing for macular dystrophy, a likely pathogenic heterozygous variant (p.Arg195Gln [c.584G>A]) was found in PRPH2 in the patient and her sister.
DISCUSSION
Different macular dystrophies, including pattern, central areolar, adult-onset vitelliform macular, and cone-rod dystrophies, as well as retinitis pigmentosa, punctata albescens retinitis, Leber congenital amaurosis, fundus flavimaculatus, and Stargardt disease, have been described in association with pathogenic variants in PRPH2. More than 90 pathogenic variants in PRPH2 are associated with retinal dystrophies, with most having an autosomal dominant inheritance pattern(4-7).
Comparison of the three patients revealed that patient 2 with AVMD had better visual acuity and milder form. Both the pathogenic variants in PRPH2 and BEST1 can cause AVMD and are the only known causes of the disease; however, they are involved in only a small percentage of cases(7). The genetic basis of AVMD is unknown in most cases. A PRPH2 pathogenic variant (p.Ile33Hisfs*12) was found in patient 2. Jacobson et al. reported this variant in three individuals of the same family presenting good visual acuity and three different clinical diagnoses (pattern dystrophy, widespread retinal degeneration with more pronounced pericentral components, and no ophthalmoscopic abnormalities)(8). Patients with variants in PRPH2 exhibit phenotypic variations; however, this variability also can occur in patients with the same pathogenic variant(4,5,7,8). In the current study patients, the p.Ile33Hisfs*12 variant caused AVMD in patient 2 with typical findings, i.e., later onset; relatively good vision; and asymmetric, bilateral foveal, or perifoveal, yellow, round-to-oval subretinal lesions.
Patient 1 was diagnosed with CD. The genes most commonly causing cone or cone-rod dystrophy are ABCA4, CRX, and GUCY2D(6). Only PRPH2 was sequenced in patient 1, who had one pathogenic homozygous variant (p.Gly208Asp), which is described for the first time as a cause of central areolar pigment epithelial dystrophy(9). Here, the variant caused CD and typical symptoms, such as decreased visual acuity, color vision defects, photophobia, and decreased sensitivity, in patient 1. His family members were not examined, but he denied any macular dystrophies among them.
In patient 3, panel-based sequencing detected a variant of PRPH2. Stargardt disease is typically associated with pathogenic variants of ABCA4(4). No pathogenic variant of ABCA4 was found in patient 3, and only one heterozygous variant (p.Arg195Gln) was noted in PRPH2. This was previously reported in a patient whose pattern dystrophy was classified as “probably damaging” based on in silico analysis determined by PolyPhen2. The absence of this variant in the population databases (1000 genomes, ExAC, and gnomAD), in silico analyses indicating its possible pathogenicity (PolyPhen2, SIFT, MutationTaster, M-CAP, and CADD), change of highly conserved residues, and two new patients with this missense variant (patient 3 and her sister) all reinforce the idea that the p.Arg195Gln variant is probably pathogenic(10).
Different pathogenic variants of PRPH2 cause completely different clinical conditions and visual impairments. Assessing the mutations of this gene make it possible to group together separate disease classifications, which was previously thought to be impossible. To conclude, molecular diagnosis enables the combination of distinct diseases caused by the same gene.
REFERENCES
1. Démant P, Iványi D, van Nie R. The map position of the rds gene on the 17th chromosome of the mouse. Tissue Antigens. 1979; 13(1):53-5.
2. Chakraborty D, Conley SM, Al-Ubaidi MR, Naash MI. Initiation of rod outer segment disc formation requires RDS. PLoS One. 2014; 9(6):e98939.
3. Stuck MW, Conley SM, Naash MI. Retinal Degeneration Slow (RDS) Glycosylation Plays a Role in Cone Function and in the Regulation of RDS·ROM-1 Protein Complex Formation. J Biol Chem. 2015;290(46):27901-13.
4. Boon CJ, van Schooneveld MJ, den Hollander AI, van Lith-Verhoeven JJ, Zonneveld-Vrieling MN, Theelen T, et al. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br J Ophthalmol. 2007;91(11):1504-11.
5. Duncan JL, Talcott KE, Ratnam K, Sundquist SM, Lucero AS, Day S, et al. Cone structure in retinal degeneration associated with mutations in the peripherin/RDS gene. Invest Ophthalmol Vis Sci. 2011;52(3):1557-66.
6. Hamel CP. Cone rod dystrophies. Orphanet J Rare Dis. 2007;2(1):7.
7. Zhuk SA, Edwards AO. Peripherin/RDS and VMD2 mutations in macular dystrophies with adult-onset vitelliform lesion. Mol Vis. 2006;12:811-5.
8. Jacobson SG, Cideciyan AV, Kemp CM, Sheffield VC, Stone EM. Photoreceptor function in heterozygotes with insertion or deletion mutations in the RDS gene. Invest Ophthalmol Vis Sci. 1996;37(8): 1662-74.
9. Kohl S, Christ-Adler M, Apfelstedt-Sylla E, Kellner U, Eckstein A, Zrenner E, et al. RDS/peripherin gene mutations are frequent causes of central retinal dystrophies. J Med Genet. 1997;34(8):620-6.
10. Alapati A, Goetz K, Suk J, Navani M, Al-Tarouti A, Jayasundera T, et al. Molecular diagnostic testing by eyeGENE: analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss. Invest Ophthalmol Vis Sci. 2014;55(9):5510-21.
Submitted for publication:
October 31, 2017.
Accepted for publication:
November 2, 2018.
Funding: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflicts of interest to disclose
© 2024 - All rights reserved - Conselho Brasileiro de Oftalmologia
![]()
 English PDF
English PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket
