
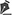
Christine Mae Morello Abbud1; Antonio Augusto Velasco e Cruz2; Harley Edison Amaral Bicas2; Fernando Chaud2
DOI: 10.1590/S0004-27492003000500025
RESUMO
Os autores relatam um caso de neurofibroma orbitário, isolado, associado à endotropia do olho esquerdo, incomitante, de aparecimento lento e progressivo há 5 anos, sem referência à diplopia. O exame da sensorialidade mostrava acuidade visual normal em ambos os olhos e ausência de diplopia devido à supressão alternante. Exames de imagem da órbita (tomografia computadorizada e ressonância magnética) revelaram lesão alongada intracônica junto ao músculo reto lateral esquerdo. A paciente foi submetida à orbitotomia para exérese de massa intra-orbitária, sem procedimento específico para a correção do estrabismo, deixada para uma segunda oportunidade. Após a cirurgia, houve diminuição do ângulo do desvio, com manutenção do quadro supressivo.
Descritores: Neurofibroma; Neoplasias orbitárias; Esotropia; Relato de caso; Literatura de revisão; Adulto; Feminino
ABSTRACT
The authors report a case of a patient who presented with a slowly progressive expanding isolated orbital neurofibroma for 5 years, associated with incomitant endotropia of the left eye, without complaint of diplopia. Sensorial examination showed a normal visual acuity in both eyes and suppression of one eye. Orbital imaging (computed tomography and magnetic ressonance) depicted an elongated intraconal lesion adjacent to the left lateral muscle. The patient was submitted to an orbitotomy for the removal of the lesion. After surgery, the eye deviation decreased without changes in the suppression pattern.
Keywords: Neurofibroma; Orbital neoplasms; Esotropia; Case report; Literature review; Adult; Female
INTRODUÇÃO
Neurofibromas são tumores benignos originados do tecido endoneural dos nervos periféricos. Histopatologicamente são formados por diferentes combinações de células de Schwann, fibroblastos e axônios(1-2). Clinicamente, eles podem-se apresentar ou como massas bem circunscritas, isoladas ou múltiplas, ou como lesões mais infiltrativas de limites imprecisos. Os neurofibromas não mostram predileção por faixa etária ou sexo(3) e podem aparecer em qualquer local, assim como no tecido ósseo(1). Um outro tumor oriundo da mesma região e com características clínicas semelhantes é o neurilemoma, ou schwannoma (também chamado de neurinoma)(2). O entendimento da anatomia dos nervos periféricos é essencial para a distinção desses dois tipos de tumores.
A maioria dos nervos periféricos é composta por feixes de fibras nervosas, constituídos por um conjunto de axônios. São três as bainhas conjuntivas que entram na formação de um nervo: o epineuro, o perineuro e o endoneuro. O epineuro envolve o nervo como um todo e emite septos para seu interior. Estes septos envolvem os feixes de fibras nervosas constituindo o perineuro. No interior desses feixes, cada fibra nervosa, ou axônio, é envolvida por uma trama delicada de tecido conjuntivo, o endoneuro. Essa camada é formada, basicamente, por fibroblastos imersos numa matriz colágena. Na maioria dos nervos periféricos, os feixes de fibras são formados por axônios mielinizados e não mielinizados. A mielina é formada pelas células de Schwann. Embora todos os axônios dos nervos periféricos sejam envoltos por células de Schwann em apenas alguns há a formação de uma verdadeira bainha de mielina(4- 5).
Histologicamente, os neurilemomas são formados unicamente por células de Schwann, enquanto os neurofibromas originam-se, como já citado anteriormente, do endoneuro como um todo(2,6).
Na órbita, esses tumores podem ser divididos em três tipos: plexiformes, difusos e isolados. O neurofibroma plexiforme geralmente está associado à neurofibromatose. Trata-se de massa não encapsulada, infiltrativa, que representa verdadeira hiperplasia das terminações nervosas, envoltas por uma camada de células perineurais(2,7). Esse subtipo é de difícil tratamento cirúrgico pois freqüentemente invade pálpebras, fossa temporal e face, apresentando tendência à recidiva(1,6,8). Os neurofibromas difusos são clinicamente indistinguíveis dos plexiformes, diferindo desses últimos pela ausência de uma distinta camada celular perineural que caracteriza os neurofibromas plexiformes(1-2).
Os neurofibromas isolados representam menos de 1% dos tumores orbitários, geralmente aparecendo da segunda à quinta década de vida(6-7,9-12). Histologicamente, mostram uma grande celularidade, variável depósito de colágeno. Geralmente, os neurofibromas isolados não são bem encapsulados, apesar do aspecto isolado e circunscrito das lesões, e são menos vascularizados do que os neurofibromas difusos e os neurofibromas plexiformes(1-2,8,12).
A malignização dos neurofibromas isolados é extremamente rara(1-2,12) e a recorrência após longos intervalos, pode ser considerado como um segundo tumor(1).
Podem aparecer em qualquer ramo neural motor ou sensorial, mas acometem mais os do nervo frontal(8). Clinicamente eles são vistos em exames de imagem (tomografia computadorizada e ressonância magnética) como lesões isoladas intra ou extra-cônicas, de margens lisas, arredondadas ou, mais tipicamente, fusiformes seguindo o eixo ântero-posterior da órbita(7,12-13). O diagnóstico diferencial inclui os já citados neurilemoma e outras massas isoladas da órbita como, os hemangiomas cavernosos, hemangio-pericitomas, fibro-histiocitomas etc(13-14).
Os neurofibromas orbitários isolados manifestam-se como massas pequenas, insidiosas, de crescimento lento, exercendo um efeito compressivo sobre o globo ocular para deslocá-lo em sentido oposto ao do tumor. Esses tumores podem ocor-rer ao longo de qualquer nervo, porém são comuns no quadrante su-perior interno ou externo da órbita(1-2,7-8,12). Do ponto de vista funcional, são lesões silenciosas que, em geral, não afetam a acuidade visual. Os pacientes normalmente se queixam de proptose lenta e progressiva quando o tumor atinge tamanho considerável(7,9). A visão dupla é o sintoma mais freqüente aparecendo somente quando os tumores são grandes e provocam restrição das rotações oculares(7,11-13,15-16).
O objetivo do presente trabalho é relatar um caso raro de neurofibroma orbitário isolado, apical, associado à endotropia, sem queixas de visão dupla.
Relato de caso
Paciente do sexo feminino, 60 anos, encaminhada ao serviço de Órbita do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto-USP, com história de endotropia progressiva do olho esquerdo há 5 anos, sem outras queixas. Na anamnese, quando inquirida sobre o seu desvio ocular, relatou ter vindo à consulta porque os familiares perceberam-no, mas ela mesma nunca o havia notado. Admitiu, apenas, diplopia ocasional há cerca de 5 anos. Na época fez tratamento para a labirintite referindo desaparecimento da diplopia. Nunca foi submetida a tratamento oclusivo ou ortóptico. Negou antecedentes pessoais e familiares relativamente a nódulos cutâneos ou manchas café-com-leite.
Ao exame oftalmológico a paciente fazia uso de correção óptica (OD: + 2,00 D esf. e OE: + 1,75 D esf., com adição de +3,00 D esf para perto), cuja adequação foi confirmada ao exame refratométrico. A acuidade visual em ambos os olhos, com correção, era igual a 6/6, sendo que na avaliação da acuidade visual do olho esquerdo a paciente apresentava posição de cabeça girada para a esquerda. Nas versões apresentava limitação importante da abdução do olho esquerdo, que atingia apenas a linha média. A pesquisa de diplopia com o teste de Maddox (filtro vermelho), foi inconclusiva pois, em condições binoculares, fixando tanto com um olho como com o outro, a paciente tinha dificuldade em perceber simultaneamente a estria e o foco luminoso (provavelmente havia supressão monocular pela alta magnitude do desvio).
A medida do desvio pelo teste de cobertura alternada e prismas revelou endotropia incomitante (tabela 1). Um desvio vertical, pequeno (D/E 4 D) foi notado, apenas, na fixação do olho direito em supraversão; por seu isolamento e baixo valor, pode ser julgado sem importância clínica.
As rotações oculares foram dadas como normais, com exceção da abdução do olho esquerdo, descrita como limitada. O quadro inicial mostra, substancialmente, uma esotropia bem maior para longe que para perto e também muito maior em levoversão do que em dextroversão. Esses dados, aliados à limitação de abdução do olho esquerdo são indicativos de paresia do reto lateral esquerdo ou de uma contenção da ação desse músculo. Por outro lado, seria de se esperar, em uma dessas condições diagnósticas, desvio maior na fixação com o olho esquerdo, quando comparado ao medido com a fixação pelo direito em quaisquer posição do olhar, o que não ocorreu (para perto e em levoversão). Ou foi pouco significativo (para longe, em infra e em dextroversão). Seria pois, também provável pensar-se numa endotropia com limitação de adução do olho esquerdo (o que reduziria o desvio em dextroversão). O deslocamento ocular (endotropia) poderia ser causado por um tumor; a contenção de adução também por ele, ou por um reto lateral inelástico.
Achou-se também discreta proptose à esquerda. O restante do exame oftalmológico (biomicroscopia, tonometria, fundoscopia e campimetria) foi considerado normal.
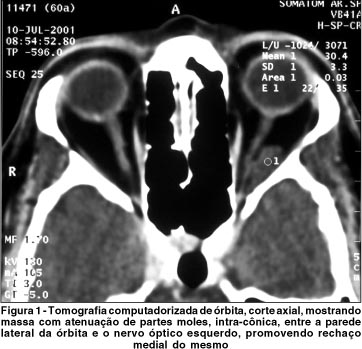
A tomografia computadorizada orbitária e a ressonância nuclear magnética mostraram massa intra-cônica à esquerda, fusiforme, de contornos bem definidos, medindo cerca de 1,8 x 0,8 cm, localizada entre o nervo óptico e o músculo reto lateral esquerdo, borrando os contornos desse músculo (Figura 1 e 2).
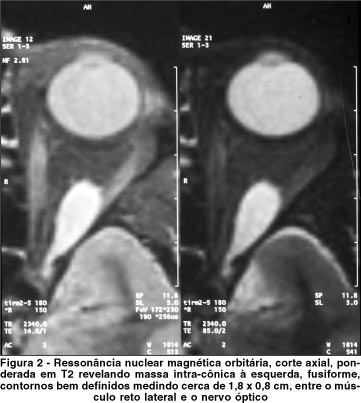
Na mesma semana, logo após concluídos os exames pré-operatórios, a paciente foi submetida a cirurgia sob anestesia geral para exérese do tumor. No intra-operatório foi comprovado, pelo teste das ducções passivas, a limitação de abdução do olho esquerdo. A técnica cirúrgica consistiu de orbitotomia lateral através de incisão coronal. O tumor apresentava-se na face interna do músculo reto lateral esquerdo no ápice da órbita. A massa foi completamente ressecada e encaminhada para exame anátomo-patológico, cujo laudo revelou neoplasia benigna intraneural constituída por proliferação de micro-fascículos nervosos em meio a fibras colágenas, observando-se diversos núcleos ondulados, com cromatina delicada e nucléolos inconspícuos: neurofibroma (Figura 3).
Logo depois da cirurgia, a paciente apresentou boa evolução, mas com piora da endotropia (ET 50 D) no pós-operatório imediato, conservando a limitação da abdução do olho esquerdo sem diplopia. Após dois meses da exérese do tumor houve melhora significativa da abdução do olho esquerdo apesar de o ângulo do desvio, em posição primária do olhar, não ter se alterado substancialmente. (Tabela 2). Este fato se justificaria com a ação do músculo reto lateral esquerdo. Um pequeno desvio vertical foi ainda notado (D/E 5D) mas apenas na fixação para longe, com o olho esquerdo. A acuidade visual manteve-se inalterada, como também o exame oftalmológico.
Num exame cinco meses após a cirurgia, valores ainda menores da endotropia foram achados (embora ela ainda se mantivesse praticamente inalterada em levoversão). Um pequeno desvio vertical (D/E 5D) foi notado em levoversão, tanto na fixação do olho direito como na do olho esquerdo (tabela 3)
Nesses exames, as condições sensoriais foram melhor investigadas. Apesar de persistir a supressão, com ausência de diplopia espontânea, pôde-se demonstrar percepção simultânea ao sinoptóforo e ao teste de Maddox com filtro vermelho, concluindo-se por correspondência visual normal. Efetivamente, chegou-se também a comprovar mecanismos fusionais muito bons (medidas do ângulo de fusão feitas ao sinoptóforo, variando entre +18D e +23D, amplitude de + 61D a -2D) e estereopsia pelo Stereo Tests®. A explicação é de que a visão binocular já existia, não havendo motivo para que ela se tornasse anômala.
Nove meses após a intervenção os desvios encontrados foram praticamente os mesmos em seus valores, resultantes de avaliações independentes de dois observadores em que as medidas respectivas das diferenças de valores achados em tais exames independentes não superaram 4D com média de 2,3D e desvio padrão de 0,8.
O exame sensorial também confirmou achados anteriores: a acuidade visual normal (1,0 com nove acertos em nove apresentações) nos dois olhos, supressão, mas com percepção macular simultânea ao sinoptóforo (ângulo de + 9D) e ao teste de Maddox (filtro vermelho). O ângulo de fusão foi igualmente + 9D, a amplitude -5D a + 38D (sinoptóforo) e a estereopsia variou entre 40" e 60" (Stereo Tests®).
DISCUSSÃO
O caso relatado apresenta achados radiológicos característicos da doença. Em especial a forma alongada conectada a um filete nervoso, acompanhando o eixo ântero-posterior da órbita. Já em relação à apresentação clínica, o caso difere radicalmente do que se espera num estrabismo de aparecimento no adulto, pois não havia queixa de diplopia (provavelmente pelo grande ângulo de desvio). Realmente, outros autores são concordantes quando afirmam que a limitação das rotações oculares é o sinal mais freqüente, principalmente elevação e abdução(13), mas que só aparece nos tumores suficientemente grandes, que provocam efeito de massa(7,11)..
O que motivou a vinda da paciente à consulta foi seu comprometimento fisionômico, pelo desenvolvimento de uma endotropia. Não é freqüente se encontrar na avaliação sensorial de um paciente adulto com estrabismo de aparecimento recente e sem ambliopia um quadro de supressão constante.
Seria possível que a paciente fosse portadora de uma microtropia antes do aparecimento do tumor. Contudo, as microtropias, em geral, não são alternantes e, portanto, provocam pequena baixa visual no olho mais comumente desviado. A paciente apresentava acuidade visual perfeitamente simétrica e normal. A possibilidade de microtropia alternante, com supressão monocular (já anterior à evolução) e descompensação do desvio pelo tumor suscitaria também, provavelmente, quadro diplópico. A outra possibilidade é de que o tumor tenha crescido tão lentamente a ponto de induzir o desenvolvimento de supressão, o que parece ter acontecido. Embora seja incomum que processos supressivos ocorram após a maturação do sistema visual, em uma extensa série de neurofibromas orbitários, publicada recentemente há menção à diplopia em apenas 12% dos casos. O autor não esclarece se a diplopia estava associada a desvio em posição primária, ou aparecia unicamente nas posições extremas, por limitação das rotações oculares(15).
Na cirurgia, o tumor foi inteiramente ressecado através de uma orbitotomia lateral com incisão coronal. Técnica cirúrgica que possibilita uma ampla exposição da fossa temporal, facilitando a retirada de toda parede lateral, incluindo a asa maior do esfenóide, além de ser totalmente estética porque não deixa nenhuma cicatriz aparente(17). Trata-se de uma abordagem clássica e, na experiência do serviço, para lesões apicais é superior aos procedimentos mais anteriores do tipo Berke, Kronlein e incisão via sulco palpebral superior.
O desvio, presente em posição primária do olhar com magnitude média (cerca de 35D), mas incomitante, com limitação da abdução no olho esquerdo, não teve tratamento cirúrgico específico durante o procedimento para a exérese do tumor orbitário, havendo sido optado por planejá-lo, eventualmente, após essa retirada. Desse modo, é possível compreender a permanência do desvio no pós-operatório imediato, aliás até com aumento, provavelmente devido ao edema e infiltração celular reativa ocasionada pelo trauma cirúrgico. De fato, houve posteriormente diminuição do desvio e da incomitância, bem provavelmente pela redução do efeito mecânico intra-orbitário da massa tumoral, fazendo com que a paciente já se considerasse satisfeita com a sua aparência, no exame de dois meses após a cirurgia (Tabela 2). Apesar de o músculo reto lateral esquerdo não ter sido ressecado com a exérese do tumor, esperava-se que a inervação do reto lateral esquerdo ficasse comprometida com a cirurgia, uma vez que a massa estava intra-cônica e justaposta à face interna do músculo. Felizmente, o desvio continuou a se reduzir, quase já desaparecendo na avaliação pós-operatória de cinco meses e de nove meses.
Seja como for, o caso ilustra claramente a necessidade de que todo estrabismo atípico seja investigado além da semiologia puramente oculomotora e sensorial. Neurofibromas apicais podem comprimir o nervo óptico e levar a defeitos campimétricos e, embora seja raro, podem sofrer transformação tornando-se malignos(1-2,12). Além disso, convém ressaltar a conduta conservadora sobre o estrabismo associado, posto que a exérese tumoral pode suscitar uma melhora de seu quadro.
REFERÊNCIAS
1. Henderson JW. Tumors of nerve sheat origin. In: Henderson JW. Orbital tumors. New York: Raven Press; 1994. p.221-37.
2. Shields JA. Peripheral nerve tumors of the orbit. In: Shields JA. Diagnosis and management of orbital tumors. Philadelphia: WB Saunders; 1989. p. 149-69.
3. Osborn AG. Diagnóstico neurorradiológico. Rio de Janeiro: Revinter; 1999. p.630-1.
4. Ortiz-Hidalgo C, Weller RO. Peripheral nervous system In: Sternberg SS, editor. Histology for pathologists. Philadelphia: Lippincott-Raven; 1997. p.285-310.
5. Scheithauer BW, Woodruff JM, Erlandson RA. The normal peripheral nervous system. In: Stout AP. Atlas of tumor pathology. Tumors of the peripheral nervous system. Washington: Armed Forces Institue of Pathology; 1997. p.7-28.
6. Shields JA, Shields CL. Atlas of orbital tumors. Philadelphia: Lippincott Williams & Wilkins; 1999. p.75-89.
7. Carroll GS, Haik BG, Fleming JC, Weiss RA, Mafee MF. Peripheral nerve tumors of the orbit. Radiol Clin North Am 1999;37:195-202, xi-xii.
8. Spencer WH. Orbit. In: Opthalmic pathology. Philadelphia: WB Saunders Company;1996. p.2629-64.
9. Krohel GB, Rosenberg PN, Wright JE, Smith RS. Localized orbital neurofibromas. Am J Ophthalmol 1985;100:458-64.
10. Meyer DR, Wobig JL. Bilateral localized orbital neurofibromas. Ophthalmology 1992;99:1313-7.
11. Lee LR, Gigantelli JW, Kincaid MC. Localized neurofibroma of the orbit.: a radiographic and histopathologic study. Ophthal Plast Reconstr Surg 2000; 16:241-6.
12. Rootman J. Neoplasia. In: Rootman J. Diseases of the orbit. A multidisciplinary approach. Philadelphia: Lippincott Williams & Wilkins; 2003. p.241-56.
13. Dervin JE, Beaconsfield M, Wright JE, Moseley IF. CT findings in orbital tumours of nerve sheath origin. Clin Radiol 1989;40:475-9.
14. Reese AB. Expanding lesions of the orbit. Trans Ophthalmol Soc UK 1971;91:85-104.
15. Rose GE, Wright JE. Isolated peripheral nerve sheath tumours of the orbit. Eye 1991;5:668-73.
16. Della Rocca RC, Roen J, Labay GR, Bedrossian E. Isolated neurofibroma of the orbit. Ophthalmic Surg 1985;16:634-8.
17. Rootman J, Stewart B, Goldberg RA. Regional approach to anterior, mid, and apical orbit. In: Orbital surgery. A conceptual approach. Philadelphia: Lippincott-Raven; 1995. p.151-290.
Endereço para correspondência
Christine Mae Morello Abbud
Departamento de Oftalmologia, Otorrinolaringologia e Cirurgia da Cabeça e Pescoço
Av. Bandeirantes, 3900
Ribeirão Preto (SP) CEP 14049-900
E-mail: [email protected]
Recebido para publicação em 03.06.2002
Aceito para publicação em 22.01.2003
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos ao Dr. Roberto Abucham.
© 2024 - All rights reserved - Conselho Brasileiro de Oftalmologia
![]()
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Scielo
Scielo
 Pocket
Pocket

{kind=link}
{kind=link}
{kind=link}
{kind=link}