
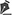
INTRODUCTION
Rothmund-Thomson syndrome (RTS) is a rare dermatosis or a condition affecting the skin. Although its exact prevalence is unknown, about 300 cases have been reported to date(1). Although its etiology is yet to be fully clarified, some patients show an autosomal recessive inheritance featuring a DNA damage repair defect and RECQL4 gene mutations(2). The diagnosis of RTS is established by clinical findings and the inability to identify a RECQL4 mutation does not rule out the diagnosis(2). The first signs of RTS are dermatological in origin. The predominant signal is poikilodermatosis, which is a patch-like pigmentation usually developing between the age of 3 and 6 months as an erythema with swelling and blistering of the face(1). Other clinical features may be present and may include congenital bone defects, short stature, sparse scalp hair, sparse or absent eyelashes and/or eyebrows, and dental abnormalities(1,3,4). Ocular findings include not only cataracts but also microphthalmia, keratoconus, corneal scleralization, and pigment deposits on the cornea and conjunctiva(1,4,5). Cataracts are reportedly bilateral, rapid in onset (usually at 2 to 3 months), and subcapsular(5,6). Several types of malignancies have been described in RTS patients, of which osteosarcoma (OS) is the most common, followed in prevalence by squamous cell carcinoma and atypical meningioma(3). Although some clinical signs suggest precocious aging, life expectancy is not impaired in RTS patients provided they do not develop cancer(7).
Case report 1
A 26-year-old man was diagnosed with RTS, with early-onset poikilodermatosis, sparse scalp hair, and dental abnormalities. He was the fifth child born to consanguineous parents after normal gestation and had an older sister and brother, both of whom were also diagnosed with RTS (Figure 1). The patient was presented to the Ophthalmology Department complaining of bilateral ocular hyperemia, photophobia, and itching for the last 12 years.
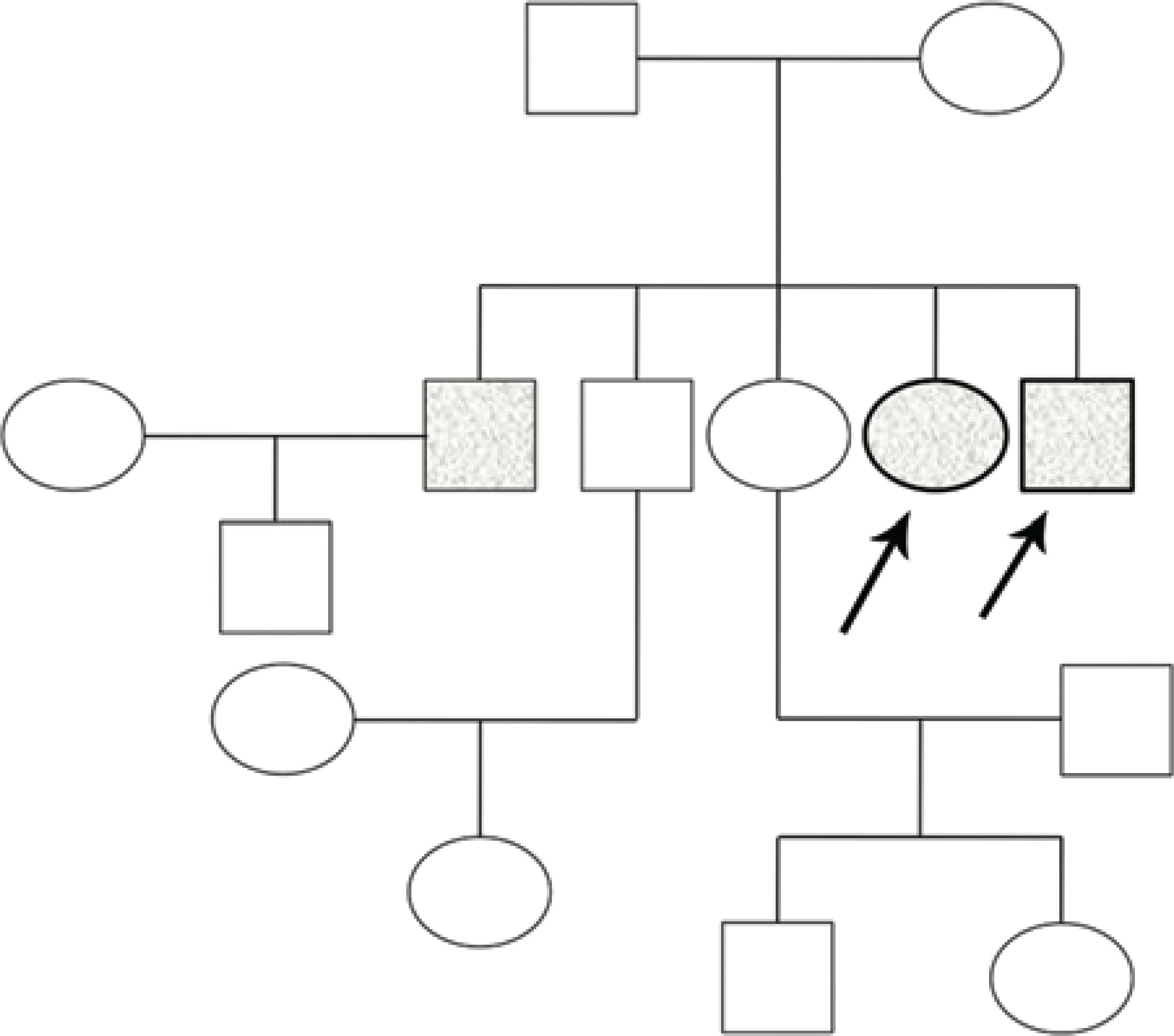
Figure 1 Family heredogram. Filled symbols represent affected individuals and unfilled symbols represent unaffected individuals; squares represent men, and circles represent women. Arrows represent the two cases.
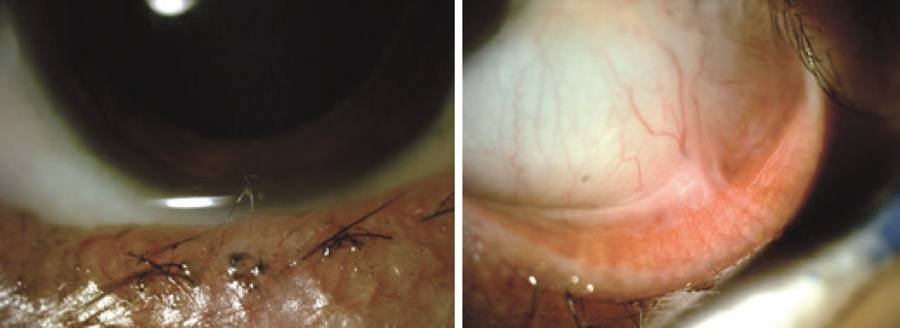
On ophthalmological examination, the patient had bilateral visual acuity of 20/20 and an intraocular pressure (IOP) of 13 and 15 mmHg, respectively. Slit-lamp examination showed bilateral bulbar and palpebral hyperemia, thickening and telangiectasia of the lid margins, inspissated orifices, secretion of the meibomian glands, and sparse inferior eyelashes, as shown in figure 2. A detailed examination of the conjunctiva revealed superior and inferior fornix foreshortening with superior and inferior symblepharon and upper lid entropion (Figure 2). Fluorescein staining showed inferior superficial keratitis. Tear break-up time (BUT) was reduced to 5 s, and reduced inferior conjunctival sac space precluded Schirmer's test. The remaining ophthalmological examination was unremarkable on both eyes.
Case report 2
The second case involved a 28-year-old woman who was also clinically diagnosed with RTS, with early-onset poikilodermatosis and sparse scalp hair (Figure 3). Five years prior to examination by the Ophthalmology Department, the patient underwent marginal rotation and lash electrolysis on her right eye; however, the symptoms persisted. She was referred for ocular hyperemia and a burning and foreign body sensation lasting for a total of 15 years.
On ophthalmological examination, the patient presented visual acuity of 20/20, and IOP was 15 mmHg on both eyes. Slit-lamp examination revealed bilateral sparse inferior eyelashes and trichiasis (Figure 4). A detailed examination of the conjunctiva led to the demonstration of inferior symblepharon on the left eye (Figure 4). Inferior superficial keratitis was evident when fluorescein staining was performed. Tear BUT and Schirmer's tests were within the normal limits. The remaining ophthalmological examination was unremarkable on both eyes.
DISCUSSION
Previous reports have established bilateral cataracts, developing early in life, as the most common ocular lesion seen in RTS patients(3), the prevalence of which varies between 5% and 73%, depending on the study(5). Absence or sparseness of eyelashes and eyebrows are also common in this syndrome(5). Other, less common, ocular manifestations appear to include microphthalmia, lacrimal obliteration, keratoconus, corneal scleralization, pigment deposits on cornea and conjunctiva, coloboma atrophy in the iris and retina, chorioretinal atrophy, congenital glaucoma, and tilted optic discs(1,4,5,8).
This report presents two cases presenting with ocular surface findings uncharacteristic of RTS, including palpebral disease and conjunctival fibrotic process. The first case presented with meibomitis, shortening of both the superior and inferior fornix as well as superior and inferior symblepharon. The signs were evident on both eyes. In the second case, bilateral trichiasis as well as inferior symblepharon in the left eye were observed. In both patients, the ocular manifestations also included previously described sparseness of eyelashes.
The first patient was referred for long-term ocular symptoms probably associated with prolonged conjunctivitis, which could explain the fornix shortening, symblepharon, and upper eyelid fibrosis with entropion. Meibomian gland dysfunction may explain the inferior keratitis, which was supported by the reduced BUT. Schirmer's test would have been important for the evaluation of tear secretion but could not be performed in case 1.
The second patient was referred for symptoms that could be explained by trichiasis, resulting in keratitis. Prolonged conjunctivitis could have resulted in inferior symblepharon. Other ocular manifestations typically described in RTS patients, including cataracts, were absent in both patients.
For both patients, the cause of inflammation remains unknown. Cicatricial pemphigoid is a possibility because some patients with ocular disease do not develop oropharyngeal, other mucous membrane, or cutaneous disease. Referring to the patients' history, we were able to exclude Stevens-Johnson syndrome and chemical injury. Considering the heterogeneity of possible clinical findings in RTS and because the two patients described here are siblings, similar and unusual ocular manifestations may also be determined by genetic factors. Histologic evaluation could have been useful but was not performed because the surgery was postponed in both patients.
In conclusion, this case report shows different clinical aspects revealed through ocular examination in RTS patients. External surface disease, particularly inflammatory and fibrotic disease of the conjunctiva, should be taken into consideration in such patients to avoid potentially severe consequences.
 English PDF
English PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Scielo
Scielo
 Pocket
Pocket
