
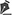
Maria Frasson1; Ana Cristina Cotta de Queiroz2; Breno Teixeira Lino1; Márcio Bittar Nehemy4
DOI: 10.1590/S0004-27492010000200016
RESUMO
Cistos vítreos são achados raros do segmento posterior ocular. Podem ocorrer em olhos com doenças oculares preexistentes ou em olhos aparentemente normais. Este estudo é um relato de caso de um paciente com retinose pigmentária e cisto vítreo, e descreve sua apresentação clínica e ultrassonográfica.
Descritores: Retinite pigmentosa; Corpo vítreo; Oftalmoscopia; Degeneração retiniana; Humano; Masculino; Adulto; Relatos de casos
ABSTRACT
Vitreous cyst is a rare condition of the posterior segment of the eye. It can occur in eyes with coexistent ocular diseases or in eyes that are otherwise normal. This study reports a case of vitreous cyst in a patient with retinitis pigmentosa and presents its clinical and ultrasonographic features.
Keywords: Retinitis pigmentosa; Vitreous body; Ophthalmoscopy; Retinal degeneration; Human; Male; Adult; Case reports
INTRODUÇÃO
Cistos vítreos (CV) são alterações raras do segmento posterior do olho, tendo sido descritos na literatura a partir do final do século XIX. São classificados clinicamente como congênitos ou adquiridos, e conforme sua cor, forma, número e ocorrência em um ou ambos os olhos(1).
A retinose pigmentaria (RP) é a distrofia retiniana mais frequente, representando um grupo heterogêneo de doenças caracterizadas pela degeneração das células fotorreceptoras da retina(2-3). Clinicamente, a manifestação funcional desta degeneração é quase sempre a cegueira legal, e as alterações fundoscópicas clássicas compõem a tríade de palidez do disco óptico, afilamento dos vasos retinianos e depósito de pigmentos em forma de espículas na retina.
Alterações vítreas na RP tem sido relatadas(4), mas o achado de CV em pacientes com RP é um evento raríssimo, sendo este o primeiro caso no Brasil, ao que se saiba, que descreve esta associação e está entre os poucos relatados na literatura.
RELATO DO CASO
Paciente de 47 anos de idade, masculino, feodérmico, foi atendido no Ambulatório de Distrofias Retinianas do Hospital São Geraldo. Havia recebido o diagnóstico de RP há cinco anos, quando procurou assistência oftalmológica devido à baixa acuidade visual progressiva em ambos os olhos (AO). O sintoma de piora visual à noite - nictalopia - estava presente desde a infância. Não apresentou passado de outras morbidades e negou traumas oculares prévios.
Seu heredograma revelou fratria de 10 irmãos (seis mulheres e quatro homens), sendo dois homens sabidamente acometidos pela RP. Um de seus irmãos foi examinado e foi constatada RP clássica, mas sem a presença de CV. Seus pais eram não consanguíneos, já falecidos, sem história de doenças oculares, mas seus avós maternos eram primos em primeiro grau.
O exame oftalmológico mostrou acuidade visual de 20/100 no olho direito (OD) e também de 20/100 no olho esquerdo (OE), com refração plana. Os reflexos pupilares, assim como o exame de motilidade ocular, estavam normais. A biomicroscopia do segmento anterior evidenciou a presença de catarata polar anterior discreta em AO, e o terço anterior do corpo vítreo se encontrava normal.
A medida da pressão ocular pela tonometria de aplanação (Goldmann) foi de 14 mmHg em AO.
A oftalmoscopia binocular indireta revelou um grande cisto flutuando na câmara vítrea do OD, na região nasal inferior, próximo ao disco óptico. Sua superfície era lisa, não pigmentada, e seu formato era oval, com a imagem se assemelhando a de um cristalino luxado no vítreo (Figura 1). A relativa mobilidade do cisto pôde ser notada com os movimentos oculares do paciente, dando a impressão de não estar preso a nenhuma estrutura do segmento posterior.
Em seu terço posterior, o corpo vítreo de AO apresentou opacidades granulares difusas, esparsas e não pigmentadas, além de pequena sinérese. O restante do exame fundoscópico do paciente correspondeu ao aspecto típico encontrado na RP: disco óptico com palidez difusa; afilamento dos vasos retinianos e presença de pigmentação retiniana em espículas, com quadro simétrico em AO. Neste paciente, foi notado um padrão paucipigmentário da retina caracterizado por uma profunda palidez retiniana, presença de poucos depósitos de pigmentos e intensa atrofia do epitélio pigmentário da retina (EPR). A região macular tinha um aspecto metálico e não apresentava reflexo em AO.
Posteriormente, o paciente foi submetido à propedêutica complementar que demonstrou campo visual manual tubular (menor que 20º) e eletrorretinograma não registrável em AO.
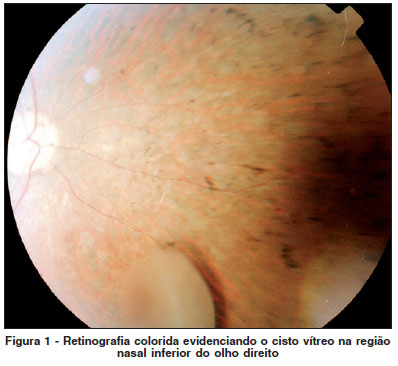
A ultrassonografia modo B do OD registrou a presença de lesão arredondada, de média reflexibilidade sonora, estrutura acústica interna pouco irregular, com cerca de 3,0 mm de diâmetros ântero-posterior e látero-lateral e 3,5 mm de diâme-tro crânio-caudal, localizada na metade posterior da câmara vítrea (Figura 2). A lesão se mostrava envolta por membranas e ecos puntiformes de muito baixas e baixas intensidades sonoras. Foi evidenciado seu deslocamento junto com os movimentos oculares, mas o cisto não alcançava a metade anterior da câmara vítrea com o paciente em ortostatismo e fletindo o mento. O restante da câmara vítrea do OD apresentava ecos puntiformes de baixa intensidade sonora, difusos.
DISCUSSÃO
O caso apresentado demonstra a forma ocasional com que a maioria dos CV é detectada, frequentemente no exame oftalmológico de rotina. O paciente não apresentava nenhuma queixa visual relacionada ao cisto vítreo no OD. Os CV geralmente são pouco sintomáticos, mas ocasionalmente determinam sintomas como moscas volantes e embaçamento transitório da visão, dependendo de seu tamanho, densidade e localização(5).
Os CV adquiridos são geralmente secundários a doenças oculares preexistentes, como é o caso da RP. Outras associações descritas são com atrofia coroideana, descolamento de retina, retinosquise, uveítes e endoftalmites(1,5). Estas doenças têm em comum com a RP a morte celular de fotorreceptores, determinando uma sobrecarga no metabolismo do EPR. Esta seria uma hipótese para a origem das alterações vítreas, inclusive os CV.
Também foi constatada a relação dos CV com traumas e inflamações oculares, embora não tenha sido comprovado que os cistos sejam consequência direta de traumas(5-6). Hipóteses como a origem infecciosa ou traumática do cisto no caso relatado são pouco prováveis, pois não houve história prévia de uveíte ou de trauma ocular.
Os CV de caráter congênito provavelmente são derivados de estruturas remanescentes do sistema vascular hialoideo, havendo discussões sobre sua verdadeira origem embrionária.
O encontro de pedículos e outros tipos de adesão membranosa nestes cistos reforça a teoria de regressão incompleta de estruturas do vítreo primário e conexões com a vasculatura hialoidea(1,6). Entretanto, a existência de cistos pigmentados levantou questionamentos sobre uma possível origem a partir dos processos ciliares ou do epitélio pigmentário iriano(7-8). Estudos sobre análises histopatológicas dos CV podem ajudar no entendimento de sua origem(6-7). Alguns autores descreveram a histopatologia de um cisto vítreo unilateral, atribuindo-lhe o diagnóstico de coristoma cístico do sistema hialóideo primitivo(9).
O pleomorfismo é uma característica dos CV. Eles podem ser ovalados ou esféricos e apresentarem superfície lisa ou lobulada, pigmentada ou não. Já foram relatadas dimensões que variam de menos de 1 mm a 12 mm, sendo que os cistos podem estar livres na câmara vítrea ou presos a outras estruturas(1,8). Foram descritos em pacientes de quase todas as idades, e as formas de apresentação também variam de múltiplos cistos unilaterais a cistos solitários uni ou bilaterais(1).
É uma condição que raramente necessita de tratamento específico, apenas quando as lesões realmente interferem na visão do paciente, ocasião em que os cistos podem ser removidos através de vitrectomia via pars plana(9).
Estudos apontaram uma alta frequência de anormalidades vítreas associadas à RP, tais como condensações, partículas pigmentadas e não-pigmentadas, degenerações fibrilares e descolamento posterior do corpo vítreo. A frequência e gravidade destas alterações não estão correlacionadas com a idade ou sexo do paciente, com a acuidade visual ou vício refracional, nem com o padrão de herança envolvido na RP(4).
Em 1952, Wagenaar investigou a associação entre cisto vítreo e RP, sugerindo a possibilidade de uma síndrome caracterizada por estas duas entidades(10). Devido à raridade desta associação, não há na literatura a descrição da frequência dos CV na RP, e embora a hipótese de síndrome seja pouco provável, as duas entidades devem estar relacionadas entre si e não serem consideradas como eventos isolados.
No presente caso, os autores acreditam que o cisto vítreo seja um cisto adquirido ao longo da progressão da RP, que se encontrava em estágio avançado. Apesar de o paciente ter tido o diagnóstico de RP há apenas cinco anos, o quadro clínico era de cegueira legal, campo visual gravemente comprometido e eletrorretinograma não registrável. Outros autores concluíram que as alterações vítreas na RP parecem aumentar com a duração da doença, acompanhando o processo evolutivo da degeneração pigmentária da retina(4). Assim como os outros tipos de anormalidades vítreas encontradas na RP, os cistos provavelmente são manifestações no corpo vítreo dos mecanismos degenerativos que ocorrem na retina e no EPR. Esta parece ser uma hipótese viável para explicar a formação do CV, mas não esclarece o motivo de sua raridade na RP, em que predominam as outras alterações vítreas.
Não foi proposto nenhum tipo de tratamento ao paciente do presente caso, pois era assintomático. Ele se encontra em acompanhamento periódico no Ambulatório de Distrofias Retinianas do Hospital São Geraldo /UFMG.
O relato deste caso enriquece a literatura por descrever uma condição raríssima e pouco compreendida. Estudos futuros sobre a RP e suas alterações vítreas poderão esclarecer a verdadeira relação do corpo vítreo com esta distrofia retiniana.
REFERÊNCIAS
1. Cruciani F, Santino G, Salandri AG. Monolateral idiopathic cyst of the vitreous. Acta Ophthalmol Scand. 1999;77(5):601-3.
2. Kalloniatis M, Fletcher EL. Retinitis pigmentosa: understanding the clinical presentation, mechanisms and treatment options. Clin Exp Optom. 2004;87(2): 65-80.
3. Unonius N, Farah ME, Sallum JM. Classificação diagnóstica dos portadores de doenças degenerativas de retina, integrantes dos grupos Retina São Paulo e Retina Vale do Paraíba. Arq Bras Oftalmol. 2003;66(4):443-8.
4. Vingolo EM, Giusti C, Forte R, Onori P. Vitreal alterations in retinitis pigmentosa: biomicroscopic appearance and statistical evaluation. Ophthalmologica. 1996;210(2):104-7.
5. Sendrowski DP, Townsend JC, Selvin GJ, Comer GW, Levoy RJ, Williams DT. Unilateral pigmented mobile vitreous cyst. J Am Optom Assoc. 1987;58(8): 656-9.
6. Asiyo-Vogel MN, El-Hifnawi El-S, Laqua H. Ultrastructural features of a solitary vitreous cyst. Retina. 1996;16(3):250-4.
7. Orellana J, O´Malley RE, McPherson AR, Font RL. Pigmented free-floating vitreous cysts in two young adults. Electron microscopy observations. Ophthalmology. 1985;92(2):297-302.
8. Bianchi PE, Guagliano R, Salati R, Traselli G. A pigmented free-floating vitreous cyst in a six-year-old child. Ophthalmologica. 1997;211(6):391-3.
9. Lira RP, Jungmann P, Moraes LF, Silveira AP. Clinical features, histopathological analysis and surgical treatment of a free floating vitreous cyst: a case report. Arq Bras Oftalmol. 2006;69(5):753-5.
10. Wagenaar JW. Vitreous cyst with retinitis pigmentosa: a new syndrome? Br J Ophthalmol. 1952;36(9):492-8.
Endereço para correspondência:
Maria Frasson
Avenida João Pinheiro, 146 - Sl. 1.303 - Belo Horizonte (MG)
CEP 30130-180
E-mail: [email protected]
Recebido para publicação em 15.02.2009
Última versão recebida em 03.10.2009
Aprovação em 04.10.2009
Trabalho realizado no Departamento de Oftalmologia do Hospital São Geraldo, Universidade Federal de Minas Gerais - UFMG.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência dos Drs. Leonardo Provetti Cunha e João Borges Fortes Filho sobre a divulgação de seus nomes como revisores, agradecemos suas participações neste processo.
© 2024 - All rights reserved - Conselho Brasileiro de Oftalmologia
![]()
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Scielo
Scielo
 Pocket
Pocket
